Geoffrey. B. Arden and David J. Ramsey
1. Introduction
There is a pandemic of diabetes. More than 350 million people are affected world-wide. In the UK more than 4.2 million people (6.3% of the population) are estimated to be living with diabetes, many without even knowing it (1). In the USA 29.1 million (9.3% of the population) have diabetes (2). Most individuals have type-2 diabetes, the onset of which is nearly universally attributed to the adoption of a “western diet”, rich in calories from refined carbohydrates and saturated fat. But type 1 diabetes is also increasing. Furthermore, type 2 diabetes now appears at much earlier ages, even in children, and increasing numbers of people with type-2 are insulin dependent (3). Individuals are not only developing diabetes at earlier ages, but also living longer. There are now various ways of controlling diabetes, but sadly these are not as successful in treating the complications of diabetes that are related to the heart, major blood vessels, peripheral nerves, kidneys and the eye(4). With an estimated 150,000 people in the UK developing diabetes each year, the prevalence of very long standing diabetes is growing even more rapidly than the prevalence of diabetes itself. This sets up a ‘perfect storm’ for the complications of diabetes which develop after long periods.
2. Classification of diabetes
Diabetes literally means “a passer through; a siphon,” so named because of the immoderate discharge of urine containing glucose which is a cardinal feature of the disease. The word comes to English by way of the Latin (diabetes) from the Ancient Greek (διαβήτης [diabêtês]). The earliest meaning of the noun “diabetes” was “compass” (5th cent. B.C.); it was not used in the sense of “siphon” (a passer through) until the 2nd or 1st cent. BC. Its first attestation as the name of a disease is in the 2nd century A.D. by Aretaeus, a Cappadocian (Greek) physician and medical writer (Figure 1), and by his contemporary Galen.

Aretæus of Cappadocia, writing in the 2nd century A.D., is the earliest physician whose account of this disease is extant: “Diabetes is a wonderful affection, not very frequent among men, being a melting down of the flesh and limbs into urine.” (5)
The first occurrence in English may be in a 1425 translation into Middle English (non vidi) of Chauliac’s seminal work on surgery, Chirurgia magna, which was finished in 1363. In seven volumes, the treatise covers anatomy, bloodletting, cauterization, drugs, anesthetics, wounds, fractures, ulcers, special diseases, and antidotes. The earliest attestation of “diabetes” in a primary English work comes from circa 1541, R. Copland’s Formularie of Helpes of Woundes & Sores in Guy de Chauliac’s “Questyonary Cyrurgyens” “Yiijv, Auycen Graunteth in diabete the water of the clere mylke of a shepe.”, closely followed by an even clearer reference to the disease in Dean William Turner’s book Natures Bathes (1562): “It is good for the flixe to the chamber pot called of the beste Physicianes Diabetes, that is when a man maketh water oft and much.”
Diabetes can further be broadly divided into two classes of disorders, diabetes mellitus and diabetes insipidus. Both share in common the production of copious quantities of urine, as well as an associated risk of dehydration and electrolyte imbalances; however, the two diseases originate from very different sources. When the term “diabetes” is used without qualification, it almost always refers to diabetes mellitus. Diabetes mellitus is a group of severe metabolic disorders marked by high levels of blood glucose resulting from defects in the production of insulin, resistance to that action of insulin, or a combination both. Greek physicians used to diagnose the condition by tasting the urine to detect saccharine matter. In 1675, the English epidemiologist Thomas Willis (1621–1675) rediscovered this phenomenon and appended the term “mellitus,” meaning “honey-sweet,” to the name diabetes (6) (Allan FM, 1953). To some extent this diagnostic practice continued into the 20th century among physicians who were taught to use literally all of their senses in the art (and science) of diagnosis. Diabetes insipidus is a relatively rare disorder and glucose does not ordinarily spill into the urine of patients, hence the term “insipidus,” Latin for “tasteless,” appended to the name (6).
3. Types of diabetes mellitus
There are two main forms of diabetes mellitus: type 1 and type 2. In years gone by, these were sometimes referred to as ‘Juvenile’ versus ‘Adult’ or ‘Insulin dependent’ versus non-insulin dependent’, but some type 1 cases develop in older age, and many type 2 cases are treated with insulin. Hyperglycemia is the hallmark of both forms of the disease, the result of a failure in insulin signaling. In type 1, diabetes results from diminished secretions of insulin because of a nearly complete loss of β cells of the islets of Langerhans due to autoimmune attack. In type 2 disease a number of defects conspire to reduce the effective secretion of insulin and its activity at its receptors in various tissues. Gestational diabetes mellitus (GDM) has been retained as a separate diagnosis category (7), and a new category called “other specific types” created to cover the growing number of cases where specific genetic defects, surgery, drugs, or other cause of hyperglycemia have been identified (8).
Type 1 diabetes
Type 1 diabetes results from the near complete loss of insulin secretion from the pancreas. Its onset is usually acute and associated with increased output of urine, associated thirst, tiredness and weight loss (the ‘four T’s’ of toilet, thirst, tiredness, and thinner). For an unknown reason, autoantibodies are produced that destroy the insulin-producing β cells of the pancreatic islets. The loss of β cells may begin years before diabetes is detected, and in some cases children with type 1 disease have a short remission of symptoms during the early stages of the disorder; in rare instances a complete remission may even occur (9). There is a strong genetic risk for the disease associated with the major histocompatibility complex (MHC) genes, especially certain human leukocyte antigens (HLA) alleles, but genes outside of the MHC have also been associated with increased risk of the disease (10, 11). The mechanism by which heredity makes the β cells more susceptible to autoimmune attack remains obscure.
Type 1 diabetes only accounts for five to ten per cent of all diabetic patients, but is the most common form of diabetes in children and adolescents. The prevalence is approximately one out of every 500 children and adolescents in the United States (12, 13), having risen more than thirty per cent within the last decade. The mean age of onset of type 1 diabetes is approximately 9 years of age and has been reported to occur at a slightly earlier age in girls (8.7±3.8 years) than in boys (9.1±4.1 years) (14). Recently, the incidence of type 1 disease is increasing in many populations (by as much as three to four per cent a year), a fact that cannot be explained by genetics alone (10). Although epidemiological studies have failed to identify a specific causal entity (15), space-time clustering of cases suggests that an infectious etiology is possible (16). In certain instances, a virus, again most likely by initiating the development of autoantibodies, may also induce type 1diabetes.
Type 2 diabetes
Type 2 diabetes is by far the more prevalent from of the disease, accounting for roughly ninety to ninety-five per cent of diabetic patients in the United States (17). The onset of type 2 diabetes is usually in adulthood and occurs decades after elements of the ‘metabolic syndrome’ appear. Type 2 diabetes usually has an insidious onset, and patients are for the most part unaware of the onset until they suffer from acute side effects of extreme hyperglycemia, or another illness which leads to its discovery (for example, repeated infections). Diabetes UK has estimated that up to 25% of patients who consult their general practitioners for other reasons may have undetected type 2 diabetes, and recommend that all patients have tests for glycosylated hemoglobin levels (Hb1Ac).
The hallmark of type 2 diabetes is insulin resistance, accompanied by variable defects in insulin secretion. In type 2 disease, insulin is usually present, but its activity is insufficient to meet the metabolic needs of the body. Initially, the β cells of the pancreas may be able to augment their secretion of insulin when faced with insulin resistance, but over time β cell output fails. This may result from either increasing insulin resistance, β cell exhaustion (degeneration), or both. Insufficient insulin activity causes patients to become “glucose intolerant”, i.e. experience abnormally elevated blood glucose levels following carbohydrate loads. Whereas nearly all patients with type 1 diabetes are aware that they have the disease because of the effects of severe insulin deficiency, roughly 6.2 million people in the United States with the type 2 disease are currently unaware that they have diabetes (3, 17). Often the onsets of symptoms are so gradual, or the effects of diabetes so mild or intermittent, that people with diabetes fail to recognize that they have a disorder of blood glucose regulation. However, if left untreated the consequences of diabetes are devastating. Together, these metabolic defects take their toll on the body, lead to a series of complications, and patients often succumb to an early death (15,18 ).
4. The effect of diabetes on the eye
Here we specifically concentrate on the damage that occurs at the level of the light-sensitive retina at the back of the eye. Already 240,000 people in the UK with diabetes have diabetic retinopathy. Visual impairment may occur from bleeding in the eye, often with accompanying abnormal proliferating blood vessels, retinal detachment from associated scar tissue, or even more commonly the leakage of fluid from damaged capillaries in the central part of the retina. This is called diabetic macular edema (DME). In the USA, DME is the leading cause of blindness in people of working age (3). In the UK although the prevalence is similar, there has been a recent increase in the incidence of hereditary retinal degeneration, so DME is now the second most common cause of registered blindness in the working age group (4).
5. Detection and screening
Once diabetes has damaged the retina, vision is often permanently lost. For this reason all efforts are being made to detect diabetic retinopathy (DR) as early as possible by taking photographs of the back of the eye (Figure 2) each year in every person who had diabetes. In the UK, for example, over 3 million people are examined in this way each year, and over 100,000 of these are referred to the eye departments of NHS Trusts, which are struggling to cope with these large numbers of patients. National eye screening programs using fundus photography and reading centers have been set up in many health care centers throughout the world.

Clinically, DR is divided into nonproliferative and proliferative disease, the latter of which is marked by evidence of new vessel formation. Non-proliferative diabetic retinopathy (NPDR) can further be divided into mild, moderate and severe, “pre-proliferative” diabetic retinopathy. The term “background retinopathy” is no longer used.
As patients with sight-threatening retinopathy may not have symptoms, life-long evaluation for retinopathy by annual screening is a valuable and necessary strategy. Pediatric patients with type 1 diabetes generally do not need to have a screening exam for retinopathy before age 9, or within 3 to 5 years of diagnosis, but should have them yearly thereafter (19). This is because DR generally does not start to develop until approximately five years after the onset of diabetes and is unusual before puberty; on average, only half of patients with type 1 diabetes develop retinopathy by eight to ten years after the onset of disease (20, 21). By contrast, because patients with type 2 disease may have had diabetes for ten or fifteen years prior to their diagnosis (22), it is essential that they are screened at baseline and yearly thereafter (23-25). Recent data from the Liverpool eye study suggest that a longer interval after an initial exam may be appropriate for type 2 patients. Type 2 patients without clinically detectible retinopathy at baseline remained free of sight-threatening DR for an average of 5.4 years (26). The complications develop insidiously often going unnoticed by patients until there is a sudden and dramatic loss of visual acuity caused for example by preretinal hemorrhages from new vessels or loss of central vision from retinal swelling. The fundus photographs (Figure 2) show small circular “dark lesions”, which are microaneurysms or “dot and blot” hemorrhages, or lighter exudates. Swelling of the macular retina is also detectable (for more details see (27)). Microaneurysms are thought to be due in part to loss of pericytes (27). In the resulting capillary dilatation, blood flow is sluggish and localised clotting may occur. In the end stages of this process the whole capillary is often obliterated. This can be seen histologically as ‘ghost vessels’ and on fluorescein angiography of the retinal circulation localized areas appear where the inner retinal capillary blood supply is lost. Dark-adapted thresholds are elevated early in diabetes (26, 28-31) and selective loss of blue–yellow colour contrast sensitivity occurs early in the disease (32-34). Clinical screening by slit lamp biomicroscopy and indirect ophthalmoscopy, complimented by fundus photography, and often optical coherence tomography (OCT) is necessary to monitor the disease in individuals. More recently, the advent of wide field and now ultra-wide field angiography have the potential to aid in the diagnosis and management of retinopathy (35, 36) but have not yet been incorporated into any screening programme.
In animals, results similar to those in man can be seen, but there are species differences. Rodents develop microaneurysms (37) but these changes are much less pronounced than in man (38) and proliferative retinopathy is not seen or the later hemorrhagic changes seen in man. These do appear in longer-lived species such as dogs. Swelling of the retina can be seen in mouse, but it is not clear how closely related this is to human DME.
The different stages of the disease are seen in the following series of retinal photographs. The progress of DR is most clearly seen in fluorescein angiographs where the damage to the retinal vessels and arterioles are more obvious than in the fundus photos. These studies also highlight regions of capillary dropout and other microvascular abnormalities.
6. Clinical grading of diabetic retinopathy and stage of disease
Nonproliferative retinopathy
In man the earliest stages of the disease, loss of retinal capillary pericytes, contributes to mechanical changes in the retinal blood vessels, altering their patency and permeability (39). These changes consist of microaneurysms (i.e. focal dilations) of retinal capillaries located in the inner nuclear layer that can be appreciated by the direct ophthalmoscope as tiny red dots about the size of a pinhead in the retina. (Isolated microaneurysms also appear in a number of other retinal diseases, including glaucoma, hypertensive retinopathy, and venous occlusal diseases like central retinal vein obstruction). These vascular changes often progress to yield hemorrhages within the retina. Hemorrhages in early DR usually appear as either “dot/blot” or “flame” hemorrhages, however, severe retinal bleeding can occur. Dot hemorrhages (tiny) and blot hemorrhages (larger) are the result of the rupture of weakened capillaries (i.e. aneurysms) within the neural retina. Flame hemorrhages are the result of the same underlying process, but occur in the superficial layers of the retina causing the blood to track along nerve fiber bundles. Hard exudates, appreciable as yellow-white spots in the peripheral retina or sometimes in a pattern of streaks around the macula, also tend to increase in number. These precipitates consist of lipoproteins and other proteins that have been extravasated from leaky retinal blood vessels as a result of the failure of the blood retinal barrier (40). Finally, microretinal infarcts often occur in the nerve fiber layer of the retina to give rise to patches of dead retinal tissue with vague borders known as “cotton wool” spots—transient findings that are no longer considered as predictive of high risk disease as many of the other vascular features discussed below.

The patient in Figure 3 has mild non-proliferative diabetic retinopathy (NDPR) and the fundus photograph at first blush looks almost normal. The changes clinically apparent are microaneurysms, some red spots of hemorrhage, and hard yellow exudates, and on close inspection early signs of ischemia. The damage to the retina in DR is most clearly seen in the angiographs where the damage to the retinal vessels and arterioles are more obvious than in the fundus photos. The fluorescein angiogram (right) shows the hemorrhages as dark spots. This photograph was taken as the dye began to fill the vessels, and a few bright spots indicate the presence of microaneurysms. In later frames of the angiogram (not shown) these cause leakage, which is one of the mechanisms by which retinal swelling is known to occur.
Pre-Proliferative diabetic retinopathy
Pre-proliferative retinopathy (PPDR) consists of a series of signs in the retinal vasculature that heralds the worsening retinal ischemia that can ultimately lead to formation of new vessels. Progression to the pre-proliferative stage of diabetic retinopathy is marked by 1: a rapid increase in the size and confluence of hemorrhages, 2: the appearance of >5 cotton-wool spots (ischemic nerve fiber layer infarcts), 3: venous dilation and beading (focal variation in the caliber of retina veins in response to ischemia), and 4: arterial abnormalities and remodeling of existing retinal capillary beds (causing intraretinal microvascular abnormalities that do not leak fluorescein). Of these clinical signs, venous beading is considered the most significant predictor of progression to proliferative diabetic retinopathy (PDR), whereas cotton-wool spots are the least significant (because they are so transient, lasting often only a few days to a couple of weeks). About half of eyes with these retinal characteristics progress to PDR within 12 to 24 months, indicating a strong need for careful monitoring at this stage. Unfortunately, no preventative or restorative treatment exists for PPDR, although anti-VEGF agents have shown some promise in clinical trials in patients with concurrent DME (41).
Proliferative retinopathy
The hallmark of proliferative diabetic retinopathy (PDR) is neovascularization of the retina at the edges of zones of capillary non-perfusion, most often near the optic disc or within three disc diameters of major retinal vessels (42). The new blood vessels that form around the optic nerve head are referred to as “new vessel disc” (NVD) and other vessels are clinically termed “new vessels elsewhere” (NVE). In some respects, the formation of new vessel can be viewed as an attempt by the retina to revascularize areas of capillary non-perfusion; however, this process does not occur in an orderly fashion and is largely unsuccessful at restoring any lost function. These new vessels penetrate the internal limiting membrane and form a dense capillary plexus between the inner surfaces of the retina or penetrate into the vitreous humor to grow along the outer surface of the vitreous body (46). The new vessels are also highly delicate and markedly permeable and may cause vision loss by intravitreal or pre-retinal (beneath the internal limiting membrane) hemorrhage. Early signs of changes in vasculature permeability can often be demonstrated by fluorescein angiography (FANG). In the late stages of the disease, these new vessels may regress, leaving only lattices of avascular fibrous tissue adhering to the retina and the posterior hyaloid membrane (42, 44). Fibroglial tissue may lead to mechanical deformation of the macula by putting traction on the retina as the vitreous body contracts, either with age or secondary to retinal ischemia (39). Retinal detachment may also occur, in some cases remaining stable for years, but other eyes rapidly progress to involve the macula and the rest of the retina, resulting in blindness if not rapidly reversed (42).
Historically, the incidence of proliferative diabetic retinopathy had been as high as 40-50% in some populations after 20-25 years of diabetes; however, the figure is now more commonly ten to fifteen per cent, nearly a four-fold decline due largely to tight glucose control that is the hallmark of modern diabetes care (43-45). However, the absolute number of new cases of blindness has not been decreasing as rapidly because of the ever expanding numbers of patients with diabetes and the earlier age of onset.
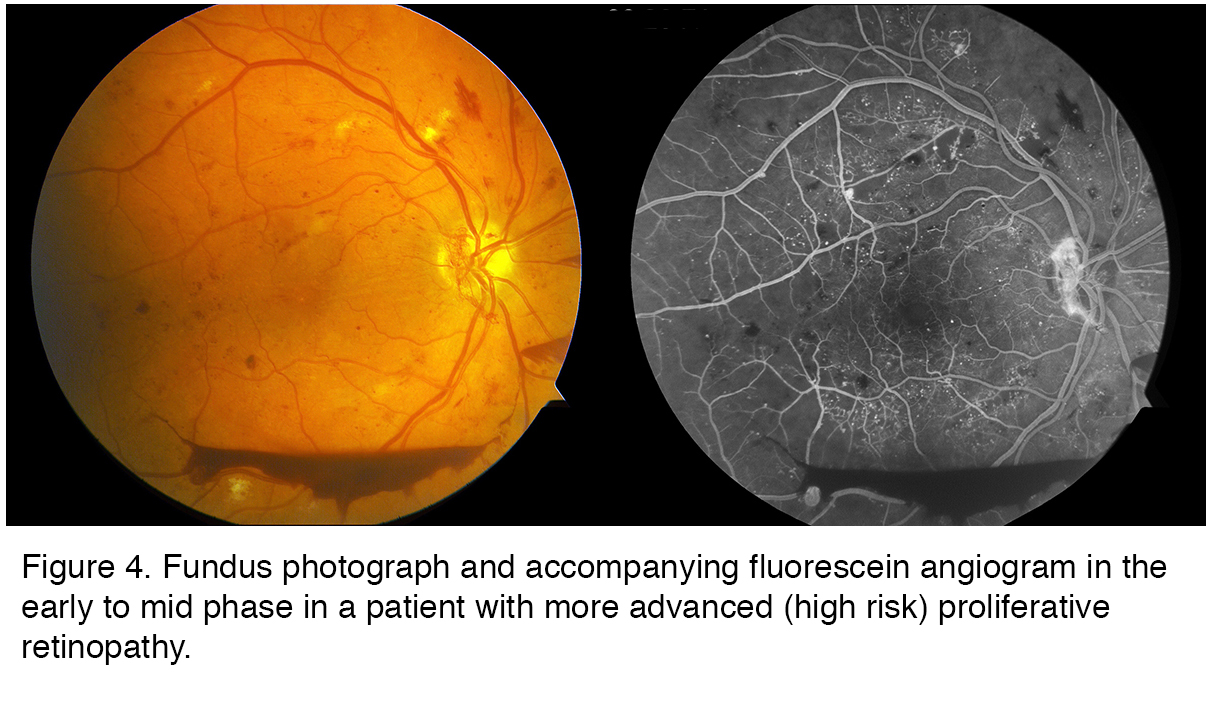
The patient whose imaging results are shown in Figure 4 illustrates many of the features of advanced PDR. There are blot and dot hemorrhages, flame hemorrhages (within the nerve fiber layer), cotton wool spots (infarcts in the nerve fiber layer), and neovascularization elsewhere (NVE) and of the disc (NVD). Inferiorly, there is a boat-shaped hemorrhage (in the lower portion of the image) that occurred some time ago. This eye has high risk features and requires treatment. The angiogram (Figure 4, right), taken at a later stage with dye filling both arteries and the veins, demonstrates leakage from the areas of NVE and NVD, as well as many more microaneurysms. Dye has leaked from the capillaries into the extracellular compartment so there is a background glow. However in some areas the capillaries have been obliterated and these areas of ischemia appear dark in appearance and are essentially dead. There is also enlargement of the foveal avascular zone (FAZ) in the central macula, which may limit central vision due to damage to the fine capillary network that services this essential region. There is also significant ischemia in the periphery (not shown).
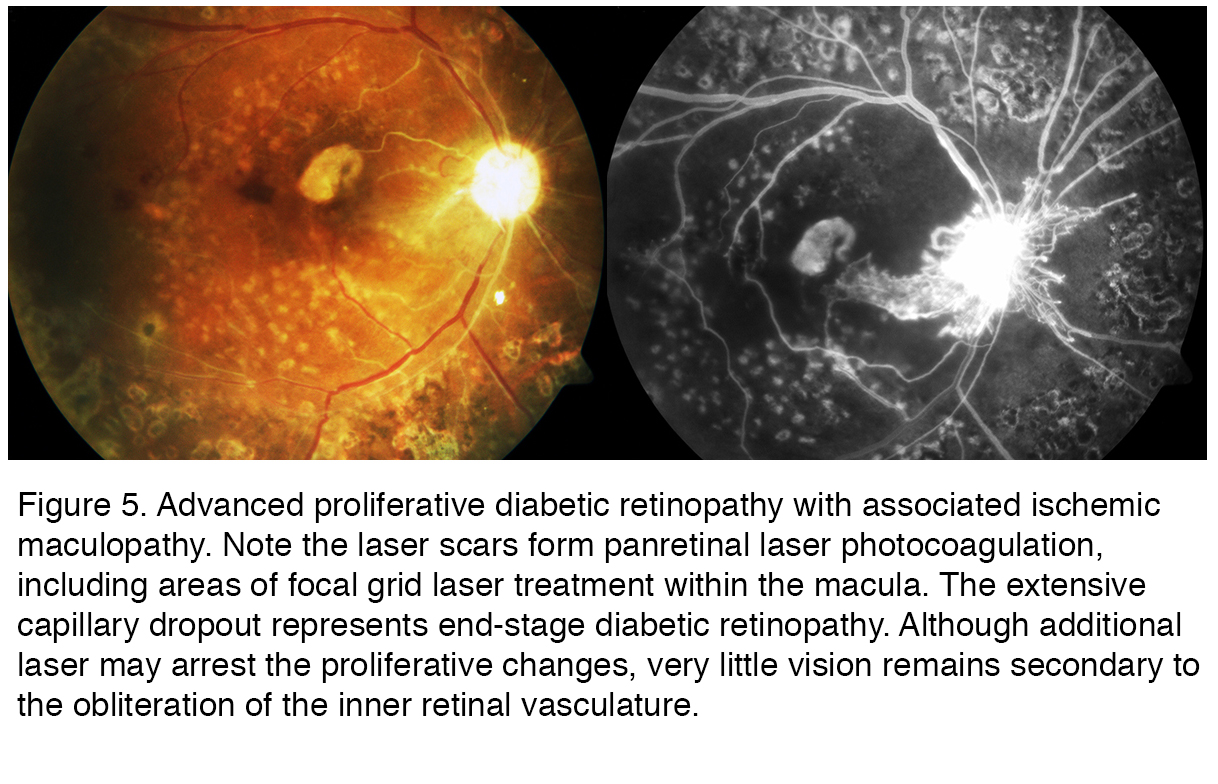
Figure 5 is from a patient with legal blindness from end stage diabetic retinopathy. This particular case is taken from a patient with type 1 diabetes who ran away from home at an early age. Living on the streets made it difficult if not impossible to handle her disease appropriately for many years. By the time she got treatment, in this case pan retinal laser photocoagulation (PRP) (seen as the scattered bright white spots in the color photography that act as window defects in the fluorescein due to the loss of pigment epithelium and overlying retina) the treatment was too late to arrest the progressive ischemic retinopathy. Ischemic maculopathy has developed, along with a central scar. Many of the larger retinal arteries and even some of the veins are no longer perfused, appearing as white ischemic vessels that remain unperfused on the FAZ. The optic disc is pale and yet new vessels emanate from the disc associated leakage seen in the FAZ. The patient unfortunately died from other complications from her diabetes in her late 20s. The progression of directly observable retinopathy often provides insight to the other complications of diabetes.
Diabetic macular edema
One of the most serious complications of diabetic retinopathy is diabetic macular edema (DME). Most of the early manifestations of diabetic retinopathy involve the peripheral retina, where changes in retinal function may be silent because of the ability of the CNS to ignore defects that develop in the peripheral visual field (46). However, as DR progresses, retinal vascular changes increase not only in number and severity, and tend to spread to the central retina where they can involve the macula, the region of the retina that underlies high acuity vision (Figure 6).
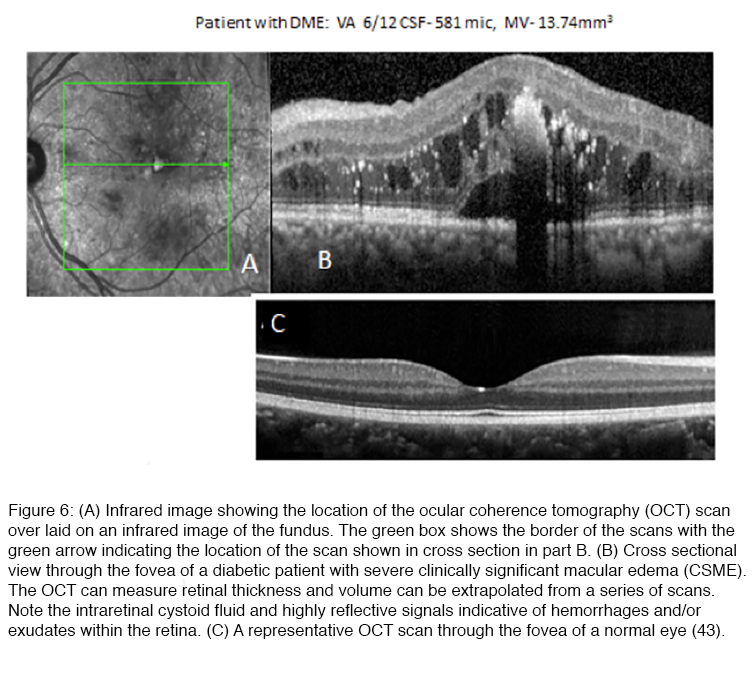
Anatomically, diabetic macular edema is the thickening of the center of the macula, i.e. fovea, due to the accumulation of fluid and plasma proteins as a result of leaky capillaries and a failure of the blood retinal barrier. Although the fovea is avascular, diabetes-induced damage to the fine capillary network that supports the function of this highly specialized region of the retina can lead to DME as the blood retinal barrier fails (40). Swelling that distorts the contour of the fovea (Figure 6), the neuroanatomical specialization at the center of the macula, can lead to a precipitous decline in visual acuity. DME usually presents as a progressive, painless loss of central vision. Patients usually report a decline in visual acuity that may develop suddenly or have a more gradual onset. DME requiring treatment may be present even when visual acuity is not yet significantly reduced (45, 46). The incidence of DME in the Wisconsin Epidemiologic Study of Diabetic Retinopathy (46-49) and the Early Treatment Diabetic Retinopathy Study (ETDRS) demonstrated that 6.4% of patients with DM develop permanent severe vision loss due to DME (44, 48-52). DME that occurs in the foveal center, if left untreated, can lead to permanent blindness (47-49). Extensive capillary closure, i.e. ischemic maculopathy, or scarring may also lead to vision loss (52). The development of optical coherence tomography (Figure 6), which uses timing or frequency differences in light reflected by different layers of the retina to examine the cross-section of the retina in real-time, has advanced our ability to diagnose and follow DME (53). Whereas “clinically significant” DME was originally based on biomicroscopic finding of macular thickening in ETDRS, many cases of DME where there are mild increases in retinal thickening sufficient to impact vision may be missed without that aid of an OCT scan (Figure 6) (54).
The standardised fundus photographs taken by screening centers (see Figure 2) have been used in attempts to grade the severity of a patient’s disease: The individual’s result is compared to a number of standardised views, by trained, registered observers whose estimates must agree. Either 2 or 7 standard areas of the retina are viewed and graded. This sort of quantitation has led to the Airlie House and the modified Airlie house grading (56, 57), which has proven value for research. The latest form of the grading appears in (27). The clinical usefulness of the classification has recently been questioned in view of the introduction of newer techniques like the OCT (55).
7. Treatment of diabetic retinopathy
1) Laser panretinal photocoagulation (PRP)
Panretinal (scatter) laser photocoagulation (PRP) is effective in treating proliferative retinopathy, reducing the risk of severe visual loss by 50% (43). Figure 7 shows the end results of such treatment. The image is a collage of different areas of retina, photographed with the same equipment as the previous figures. Care is taken to avoid PRP in the central region or macula, as this would damage the central vision, although focal laser applications are sometimes used within macula to treat areas of leakage.
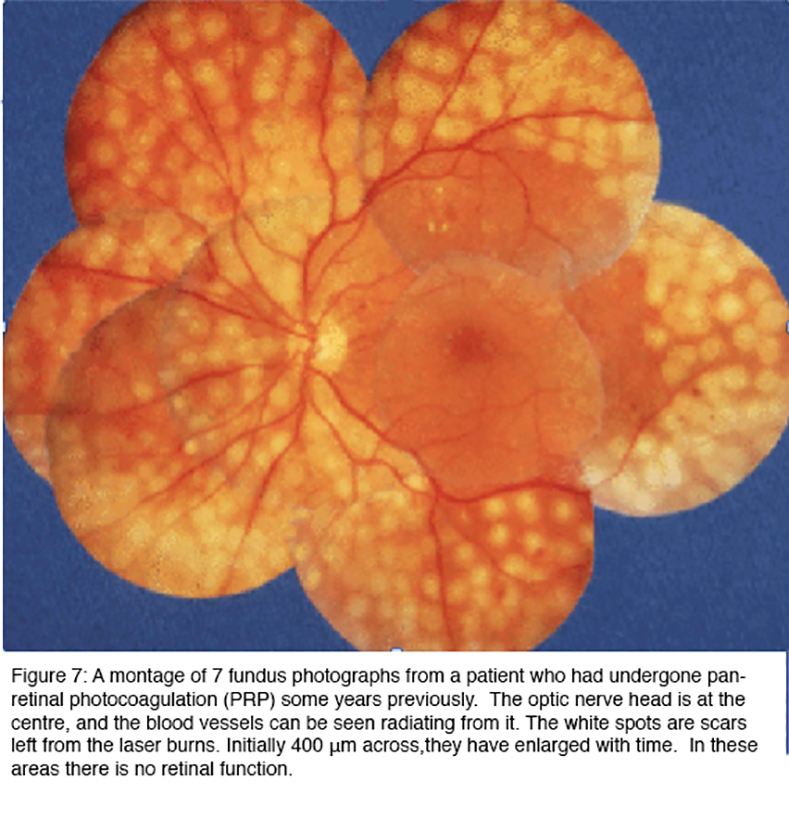
The white areas in Figure 7 are full thickness scars, in which the retina is dead. At first it was thought that the laser treatment prevented injurious substances leaking from those parts of the retina affected by diabetes (56, 57), but in fact, the burns can be placed anywhere in the peripheral retina and the treatment is successful in arresting progression of the disease as long as a sufficient area of the retina is treated. But the outcome is far from perfect. The treatment is often given after loss of visual acuity has already occurred, and it does not act to restore vision. Indeed after 5 years, slight further deterioration of acuity is the rule, although vision is conserved in patients who receive treatment compared to those who forgo PRP (58-60). Furthermore, the destruction of the peripheral retina has as an unavoidable side effect, loss of peripheral vision: and patients often complain of decreased night vision and reduced colour vision. Furthermore, the retinal nerve fiber layer in eyes that have been treated with PPR are thinner with abnormal cupping, but should not be mistaken for glaucomatous optic neuropathy (61).
2) Focal laser
The ETDRS demonstrated a 50% reduction in moderate visual loss (≥ 15 letters) in eyes with mild to moderate nonproliferative diabetic retinopathy and “clinically significant” DME (62). While a small percentage of eyes treated with laser photocoagulation had improvement in visual acuity, most did not and with time lesions often developed into focal areas of chorioretinal atrophy or scarring.
The superiority of intravitreal treatment with anti-VEGF agents, specifically ranibizumab monotherapy over focal laser alone has been noted for DME (63). In the treatment of DME, laser photocoagulation may still be used as an additional, or “rescue”, treatment. For example, in the case of the RISE and RIDE trials (identical, double-masked, sham-controlled, multicenter Phase III trials evaluating the impact of monthly ranibizumab injections on DME) laser was applied if it was found that central foveal thickness was >250 µm or if there was a 50-µm worsening from the prior month.
Recently, the development of subthreshold photocoagulation promises to greatly reduce the risk of structural and functional retinal damage from laser treatment, while retaining the therapeutic efficacy of conventional laser treatment (55, 56, 60 64, 65). The development of the diode laser with micropulsed emission with a longer wavelength in the near-infrared (810 nm) greatly reduces the thermal diffusion to the neurosensory retina and allows focal treatment without a visible burn endpoint (61). Although no retinal damage is detectable at the time of treatment, changes from the laser treatment, i.e. scarring, still develops albeit in a smaller proportion of patients compared to conventional green laser photocoagulation (64, 65, 66).
3) Intravitreal injection of medications
The situation has been transformed over the last decade with the introduction of intravitreal agents that target vascular endothelial growth factor (VEGF), one of the key cytokines whose abnormal expression has been strongly associated with the progression of diabetic eye disease. This discovery arose from an appreciation that angiogenesis of the retina of new-born animals was related to the development of local hypoxia in the retina driving the production of VEGF (67, 68). Soon after, increased VEGF expression was recognised in animal models of diabetes (69-71), as well as in samples of vitreous taken from patients operated upon for the complication of proliferative diabetic retinopathy (72). While awaiting FDA approval for ranibizumab (Lucentis), a humanized small antibody fragment (Fab) with a high binding affinity for human VEGF, was specifically designed for intraocular use (73, 74). With a smaller size compared to its parent molecule bevacizumab (Avastin) (75, 76), ophthalmologists began treating neovascular AMD with off-label bevacizumab (76). This has since become a widely adopted as a lower-cost alternative to ranibizumab. A new drug, aflibercept (Eylea), which targets all VEGF isoforms and PEGF, has been recently approved for the treatment of DME (77, 78). Ongoing studies indicate that these and related compounds may actually slow the progression of diabetic retinopathy (79). Furthermore, they are highly effective therapies for DME, often resulting in an increase in visual acuity, although some patients do not respond and other have such extensive damage that vision is permanently lost (80-85). Unfortunately, the effects are also too often short lived and the effects of diabetes progressive, necessitating repeat injections of these very costly medications (86). Delivery methods for sustained release of anti-VEGF therapy are under development (87). DME may also be treated with intravitreal glucocorticoids, particularly triamcinolone acetonide, dexamethasone, and fluocinolone acetonide.
Beginning in the late 1990s, intraocular injection of triamcinolone acetonide was proposed as an adjunctive treatment of DME and numerous clinical trials for DME followed (88). In addition, in certain cases of DME that have proved refractory to anti-VEGF treatment, steroids may provide some benefit (89, 90). In addition, sustained release devices are available and approved from some ophthalmic indications, including diabetic macular edema (91). Although these medications can be effective, the risks associated with intravitreal administration of steroid medications, especially cataract formation and ocular hypertension or glaucoma, need to be taken into account.
So the list of available treatments is short, and the results and access to treatment are not particularly inspiring. But the important message of this paper is that it is almost certain that an entirely new method of treatment, which is not only non-invasive and without side effects, but is also so inexpensive, and simple you can ‘do it yourself’ has been discovered. Below the rationale and the supporting evidence is given.
8. Pathogenesis of diabetic retinopathy
Pathological features of diabetes can broadly be divided into acute metabolic problems and long-term complications stemming from hyperglycemia-induced dysfunction, broadly divided between those that affect the macrovasculature versus the microvasculature. The general metabolic problems associated with diabetes are largely due to: (1) decrease in the proper utilization of glucose, leading to high blood glucose levels that result in an osmotic diuresis as glucose spills into the urine; (2) abnormalities in lipid metabolism, leading to the deposition of cholesterol in arterial walls and atherosclerosis; and (3) increased ketone production, with the associated risks of diabetic ketoacidosis when insulin signaling is severely depressed for long periods of time.
The most pronounced and often-cited effect of diabetes is a chronic rise in blood glucose levels. However, it is very unlikely that elevated blood glucose plays a singular causative role in the development of diabetic complications; many other cellular metabolic parameters are no less seriously altered by a lack of insulin signaling.
9. Biochemistry of diabetic retinopathy
Investigations of hyperglycemia on cellular metabolism have demonstrated that cells and tissues cultured in added glucose, or animal models made hyperglycemic by the injection of streptozotocin, experience both active and passive damage to their biochemical machinery. Increased concentration of available glucose triggers abnormalities in a range of metabolic pathways that collectively leads to “oxidative stress” as the concentration of a group of energetic radicals increases (92-94). Another line of enquiry relates to the formation of advanced glycosylated end-products (AGEs), which are modifications of proteins or lipids that become non-enzymatically glycosylated when exposed to aldose sugars and oxidative stress (95, 96). The formation of AGEs degrade the function of cellular enzymes of metabolism, as well as those that serve antioxidant functions, further contributing to oxidative stress. AGEs also contribute directly to the generation of reactive oxygen species (ROS) as a result of activating cellular mechanisms designed to rid the cell of damaged macromolecules.
Oxidative Stress
Oxidative stress occurs when the concentration of a group of energetic radicals increases (92-94). These are known collectively as ROS. ROS may include singlet or triplet oxygen, but the major contributor is the superoxide anion O2●−. In addition, in the presence of nitric oxide (NO), the oxygen radicals can react to form nitroso-compounds, but although these have slightly differing properties, and engage in different chemical reactions to the superoxide (97) this is ignored in the treatment that follows. Active species have short lifetimes and the superoxide ion rapidly transmutes into hydrogen peroxide (H2O2). The lifetimes of all these molecules depend very greatly on the local reducing environment. In cells they are surrounded by compounds and enzymes that quench them very effectively so that a sustained rise in the concentration of ROS requires either an overproduction or a reduction in the rate of their destruction. This is the case with diabetes where the production of ROS increases simultaneously with a decrease in the ability of endogenous quenchers.
Mitochondria produce ROS
Of the ways in which ROS can be produced, the mitochondria are the most important (97, 98). ROS are produced by the normal operation of mitochondria producing ATP from oxygen and trioses derived from glucose (99-103) as part of the process of oxidative phosphorylation (Figure 8). In many types of cell increased blood sugar levels in diabetes lead to increased intracellular glucose, which enters via membrane glucose transporters (104, 105). Increased glucose concentrations drive mitochondrial oxidative phosphorylation, limited by the availability of oxygen, to produce ROS (92 99-103). Figure 8 diagrams the processes occurring in the mitochondrion. Because of the short half-life of ROS, it is common to measure their activity indirectly using reporter dyes, molecules that are chemically altered by oxidation (106, 107).
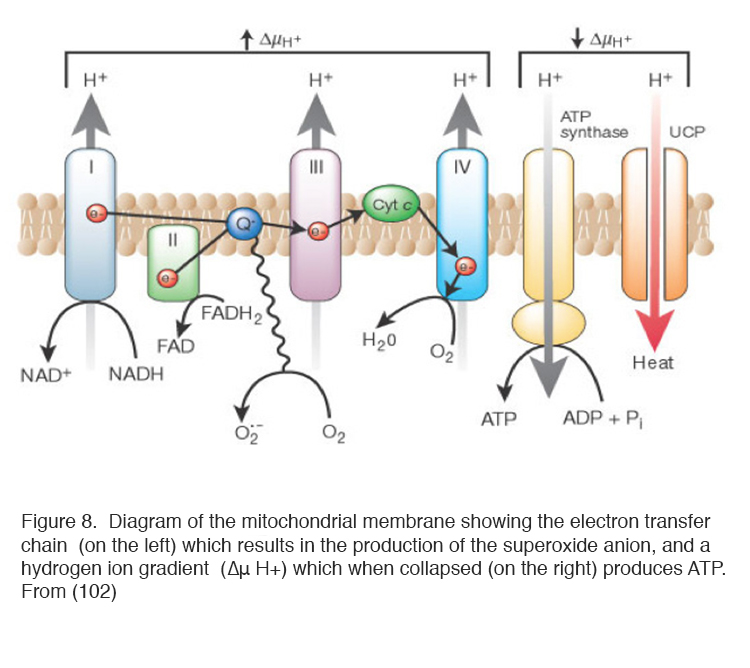
It is now possible to introduce fluorescent proteins into the mitochondrial genome and observe the process of ROS production in real time. Confocal microscopy of reporter proteins transfected into cells has demonstrated that ROS are produced in localised places on the mitochondria, where electrons leak from the electron transfer chain (ETC) in brief pulses (107). About 5% of the oxygen is diverted into this pathway and modern techniques have shown that they are emitted as discontinuous flashes (146).
The plasma membrane produces ROS
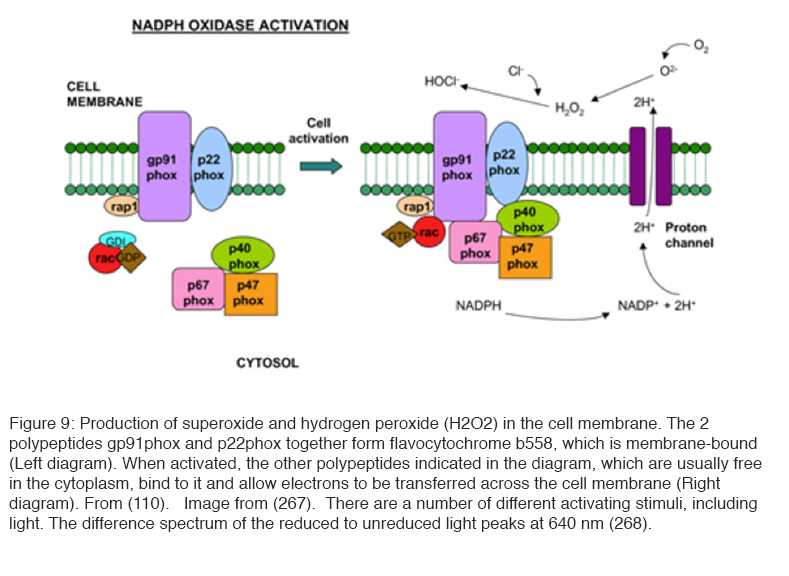
Another method of production of ROS is situated in the cell membrane and involves the enzyme NADPH oxidase (109) (Figure 9). Its activity is controlled by a complex regulatory system that involves the G protein Rac (109, 110). In resting cells, a membrane-embedded heterodimer of two polypeptides (p22phox and gp91phox)—which also contains two heme groups and copper as well as a flavine adenine dinucleotide (FAD) group—enables the transfer of electrons from cytosolic NADPH across the membrane to molecular oxygen without NADPH oxidase activity. It is believed that the charge compensation occurs when gp91phox polypeptide also acts as a proton pump. Upon stimulation, a number of polypeptides (p47phox, p67phox, and p40phox) translocate to the inner face of the plasma membrane to form a fully active enzyme complex that contains NADPH oxidase activity. The central polypeptide gp91phox also acts as a Ca2+ or H+ channel, and the ROS produced appear to regulate signaling pathways (109, 110).
Targets of oxidative stress
Most ROS are formed in a highly reducing environment inside cells, and must pass through cell membranes into the extracellular environment (which also contains quenchers) before they can diffuse to other cells. Oxidative stress makes membranes more permeable (111, 112), and reduces the quencher concentrations. But initially, in the early stage of diabetes, ROS are unlikely to travel more than a few microns, lasting for very short periods, before being captured and quenched, and therefore the earliest damaged in diabetic retinopathy occurs because of changes initiated intracellularly (113). Morphological evidence of intracellular oxidative stress is the swollen and disrupted cristae of mitochondria (114). Evidence of oxidative stress can be seen in many of the layers of the retina, for example the inner nerve fiber layer and ganglion cell layers, and this recently has been correlated with changes in the OCT (115). Localised oxidative stress in rodent retinal capillaries appears to develop rather late. Most of the ROS production however is in the photoreceptor layer (116). Although photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation, the functional disturbance to the phototransduction process is minimal, limited to the recovery rate after intense bleaching, and the responses to threshold stimulation (117-121).
Caveats to the oxidative stress theory
Despite this evidence, the precise mechanism by which hyperglycemia leads to the increased concentration of ROS is uncertain. In humans, there is no simple relationship between hyperglycemia and the development of retinopathy (122-124) (except for the very highest levels of hyperglycemia), so the determining factor may be when hyperglycemia reduces the capacity of antioxidant defences (125, 126). Experiments on tissues in raised glucose are usually confined to comparing the results in 5mM with 25 or 30mM glucose. In one case (studies implicating increased glucosamine with oxidative stress) (127), the threshold appears to be about 12mM. This corresponds to the highest levels of hyperglycemia found in normally functioning patients (122-124). At the glucose levels utilized in most biochemical investigations in excess of this level, patients would soon be severely ketotic or comatose with hyperosmolar non-ketotic decompensation syndrome, a condition that carries a mortality of 40% (128). It is therefore difficult to be sure that the ‘downstream’ abnormalities demonstrated in laboratory experimentation in “high” glucose have key roles to play in the development of retinal lesions, which develop in patients with relatively better controlled hyperglycemia. Rodents made diabetic with streptozotocin have body weights, which are up to 40% less than age-matched “controls”, and results obtained under these conditions are possibly not directly applicable to clinical situations either. In fact, oscillating levels of elevated blood glucose levels appear to be more damaging than persistently elevated levels (129). Hyperglycemia can only have a direct effect if it results in higher intra-cellular glucose. While glucose can penetrate some cell membranes, in most it is ferried by glucose transporters (130), and either the number of portals increases or each turns over faster. It is of interest that in human retinal endothelial cells grown in monoculture, glucose transport does not increase under conditions of excess glucose, intracellular hyperglycemia does not occur, and the cells in culture do not show signs of oxidative stress (130, 131). This leads to the conclusion that the damage to capillaries by ROS (which are fundamental to the development of diabetic retinopathy) is due to a paracrine mechanism, i.e. they are secondary to some less direct effects caused by hyperglycemia.
Polyol pathways
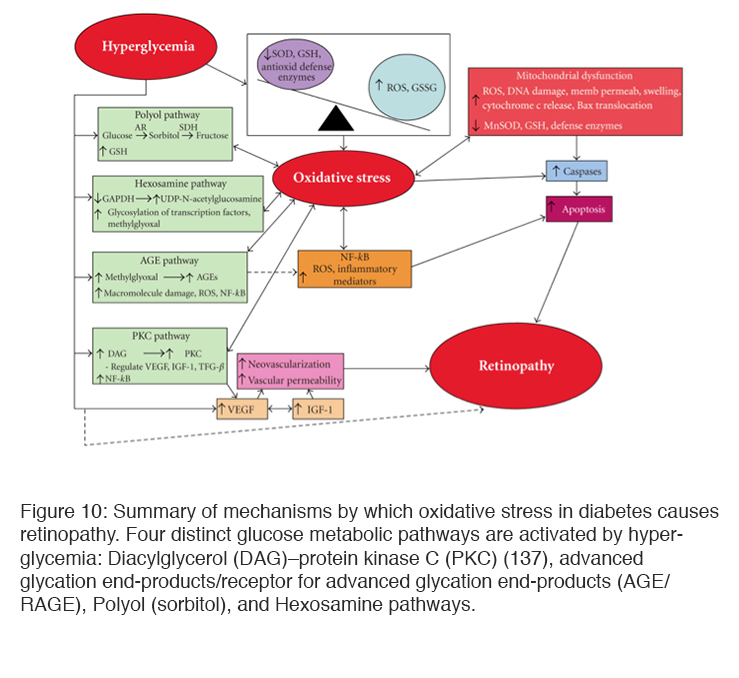
Excess glucose not consumed by the tricarboxylic cycle may also be shunted to glycolytic pathways or enter the polyol (sorbitol) (133, 136) and hexosamine pathways leading to a build of up of metabolites that damages cellular function. Protein Kinase C (PKC) has also been found to be elevated in hyperglycemia. Increased glycolysis leads to the synthesis of diacylglycerol (DAG) which is a key activator of PKC. PKC in turn increases endothelial permeability, the production of VEGF, and leucostasis as well as endothelial cell proliferation (134, 135). Sorbital build up has been found in many cells (including retinal pericytes from diabetic animals). The reaction involves oxidation of glucose by NADPH, and this can cause a reduction of glutathione, one of the reducing agents that remove ROS (132), and increased accumulation of sorbitol is also thought to cause osmotic damage (133). Activation of these metabolic pathways leads to microvascular dysfunction, neuronal apoptosis, glial reactivity and inflammation (137, 138) (Figure 10).
Mitochondria also respond to hypoxia. The first effect is to reduce the formation and release of ROS (139, 140): However mitochondria and other cell-surface mechanisms, also initiate signaling cascades that increase ROS (141-143, 144-152). The effect of alteration of the ROS output is however uncertain because change in pO2 can also increase the output of quenchers, (148) at the same time as ROS are increased.
Advanced glycosylation end products
Hyperglycemia also results in the formation of complex molecules known as advanced glycosylation end products (AGEs) that affect the development of oxidative stress in several different ways (148) (Figure 11).
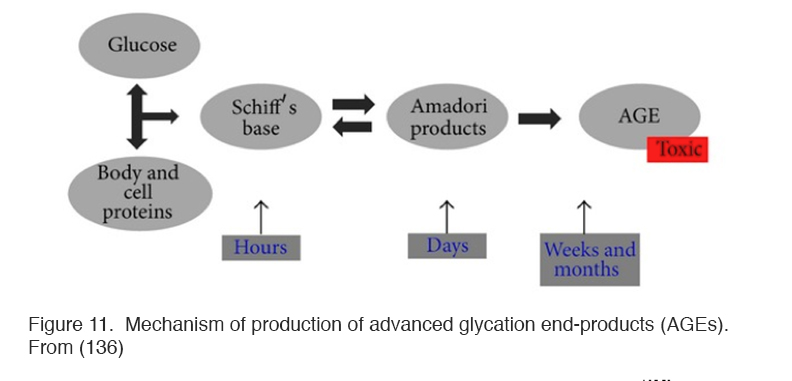
Figure 11. Mechanism of production of advanced glycation end-products (AGEs). From (136).
AGES contribute to the thickened basal membranes found in diabetes, which hinder diffusion of oxygen and nutrients both from blood to extracellular fluid and also within tissues. In additions, there are smaller molecule AGEs which interact with a receptor named RAGE, found on Müller and other cells, which, when satisfied, contribute directly to oxidative stress by directly affecting the NADPH oxidase system (149). RAGE also causes apoptopic cell death of retinal neurones by a mechanism involving the overproduction of nitric oxide (150). AGEs also directly affect retinal blood vessels causing leakage which is associated with increased VEGF and VEGF mRNA expression (95, 96, 150). The immediate effect of hypoxia is to reduce the production of ROS, as would be expected as the metabolism of the mitochondria slows (150). But part of the mechanism of production of ROS, the NAD/NADH ratio is changed by AGEs, producing “pseudo hypoxia”, and this change increases oxidative stress (109, 144, 147).
The search for treatments
Many of the pathways involved in producing oxidative stress-induced cell death have been investigated by using selected animal models in which the activity of a particular pathway is increased. If the same experiments are carried out in animals in which quenchers are also increased, oxidative stress is reduced or may not appear (152, 153). If the stimulus driving oxidative stress is removed (e.g. hyperglycemia reduced) biochemical changes may not be restored to normal. It has been shown that ROS and reactive nitrogen species (RNS) activate a spectrum of transcription factors, including nuclear factor kappa B (NFκB), AP-1, p53, Ets transcriptional factor (Ets-1), hypoxia-inducible factor-1α (HIF-1α), and Nrf2. (154, 155) and following reversal of hyperglycemia to normoglycemia the transcription and regulation of proteins remains affected for months. This has been described as ‘molecular memory’ and due to changes in the nucleus. Damage to mitochondrial DNA also occurs(151, 155), and variations in mitochondrial DNA are associated with different probabilities of development of severe diabetic retinopathy (156) related, at least in part, to the expression levels of particular proteins. Amongst these is the expression of Vascular Endothelial Growth Factor (VEGF), interleukin 2 and transforming growth factor (TGF). All this experimental work has provided a number of blocking agents which can reduce the production of oxidative stress in laboratory conditions (157-171), but almost all have had inconclusive results in subsequent clinical trials (171-176). This has been explained (155) by the hypothesis that so many biochemical pathways contribute to and result from oxidative stress that blocking one is relatively ineffectual (108, 177).
Cytokines
The relatively spectacular improvement seen clinically with anti-VEGF drugs emphasises the importance of this cytokine as a final common pathway of damage to the retina and its vasculature. In turn the damage caused by excess VEGF prompts the production of abnormal concentrations of other related growth factors. Another observation quite unexplained, is the finding that in dogs, after 5 years of hyperglycemia, pericyte loss and capillary dropout occurs in retinal capillaries but not in brain (178): there appears to be a specific retinal cause for diabetic retinopathy, although the two tissues have the same neuroectodermal origin.
10. Neuronal abnormalities in early diabetes
In addition to the biochemical changes discussed above, which may cause the clinical vasculopathy, it has been known for many years that retinal neuronal damage occurs early in the condition (179). The examination of human post-mortem material (180) showed loss of tissue and excess TUNEL staining characteristic of apoptosis (181, 182). Thinning of the retina in living diabetic patients has been described (183). However the damage is selective: Ganglion cells and short wavelength cone receptors are preferentially damaged (184). Experiments on streptozotocin-induced diabetes in rodents (185) have led to similar claims. Thus more apopotosis has been claimed by some (186, 187), but has been reported absent by others (188). An example of damage to astrocytes and disappearance of connexin-43 normally seen in association with astrocytes is shown in Figure 12 (187). Electrophysiological testing (189-191) both in man and rodents indicates inner retinal functions are disturbed much more than outer retinal function (31, 191). In psychophysical testing, losses in tritan colour vision, originally thought to be due to scattering changes, have been shown to be neural, to occur very early, and to be ameliorated by oxygen inhalation (192). Similar aberrations are seen with other psychophysical measures (189, 193-196).
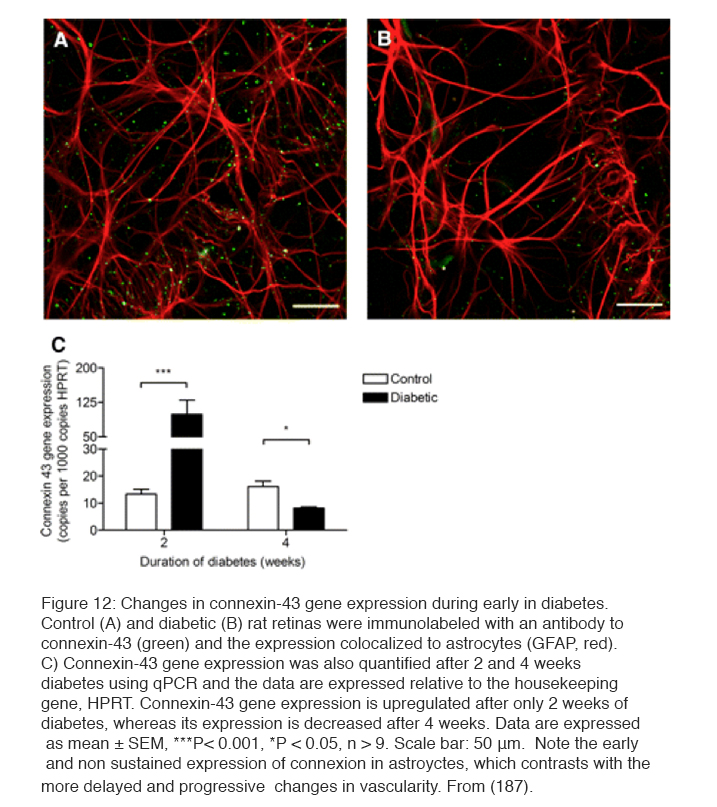
There is some doubt about whether the neural changes precede the onset of the vasculopathy (197-198). Most biochemical experiments are studies after >4 week period of diabetes (216). Hypoxia and inner retinal neural changes have been reported at 2 weeks and the earliest vascular changes (leakage of fluid from retinal capillaries) can only be seen after 4 weeks. The early neuronal changes (187, 197, 198, 200) also occur in the absence of changes in the ERG which are dependent on the outer retina (201) (and see below). So it is possible that vascular and neuronal changes in the rodent retina are produced by different mechanisms, just as may be the case in man.
In superfused retinal slices, patch clamp recordings have demonstrated changes in glutamate release and activity in the rod pathway. These changes in spontaneous excitatory postsynaptic currents (spEPSCs) are an indicator of retinal neuronal activity and are controlled by hyper-glycemia. Thus excess activity in spEPSCs can be seen in the rod bipolar terminals recorded in slices from the retinas of rats made diabetic with streptozotocin (202). The activity returns to normal when the superfusing saline contains less glucose. The rod bipolar cell has a metabotropic glutamate receptor at its synapses with rod photoreceptors, and becomes depolarised on light stimulation. It releases glutamate at its axonal synapses with AII and A17 amacrine cells (see chapter in Webvision on Rod pathways through the retina). The glutamate release is increased in diabetics and returns to normal if the superfusing fluid does not contain extra glucose. The glutamate affects the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) receptor in A17 cells. This, by affecting [Ca2+]in , reduces the inhibitory GABAc receptor activity at the rod bipolar cell and A17 synapse, and, in turn, still further increases the glutamate output by the rod bipolar cell (Figure 13) (202) (see chapter in Webvision on GABAc in the retina).
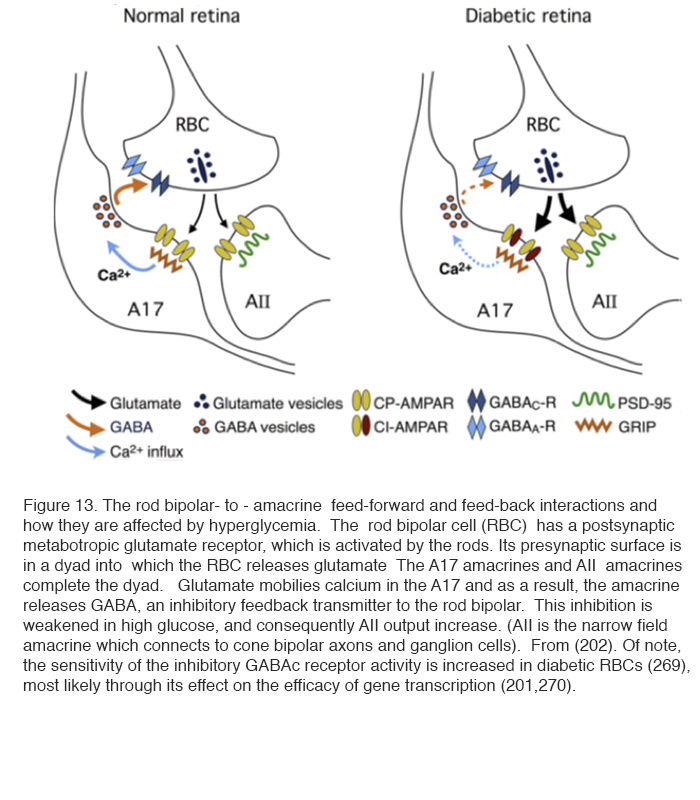
Thus not only does there appear to be a specific retinal cause for diabetic retinopathy (178), but also this cause is linked to hypoxia and metabolic overload, both in neuronal and vascular retinal elements. This was the trigger for the new hypothesis discussed below.
11. A new hypothesis based on rod photoreceptor biophysics
Hypoxia, driven by dark adaptation, accelerates the development of diabetic retinopathy (189, 203-205). There are several reasons why this occurs: Rod photoreceptors in the outer retina are maximally depolarized in the dark and continuously release large amounts of their neurotransmitter glutamate and this increases energy utilization and oxygen consumption (206-208) (Figure 14).

In darkness rods consume more oxygen than any other cell in the body (208-210) because the water and ions entering the rod outer segment need to be pumped out by pumps localized near the large mitochondria of the inner segment (Figure 14) (this pumping has similarities to a fireboat squirting out extreme amounts of water, Figure 15).

Even though the choroidal blood supply is very profuse, the partial pressure of oxygen at the level of the inner segments of the photoreceptors is near zero in darkness compared with the 30 mm Hg common in most tissues. In animal experiments, the partial pressure of oxygen rises suddenly and briefly when the retina is stimulated by a flash of light (211). With normal oxygen concentrations, the rods receive sufficient oxygen, but if oxygen is reduced somehow, the rod sensitivity falls. This occurs with only a modest reduction of oxygen corresponding to a reduction in air pressure that occurs at an elevation of about 4000 ft. Thus, in commercial jet aircraft, which are pressurised to around 7000 ft., healthy normal observers have slightly reduced rod sensitivity and some individuals, particularly those with heart or lung disease, may experience symptoms of dimmed vision, fatigue, nausea, headaches, loss of consciousness, and (on extended flights) even pulmonary edema or death from altitude sickness (212). Consequently, if in retinal disease the availability of oxygen is reduced due to decreased function of the retinal circulation, or the relative demand for oxygen increases due to metabolic stress, there will be an imbalance which will lead to hypoxia in not only the outer retina (which is avascular) but also in the inner part, which is supplied primarily, but not completely, by the retinal arterial circulation. These processes are diagrammed in Figure 16.
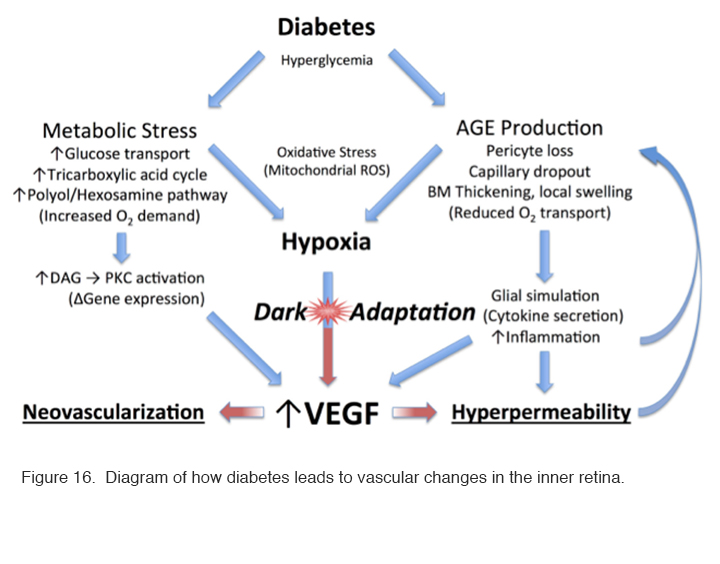
Figure 16: Diagram of how diabetes leads to vascular changes in the inner retina.
Increased blood sugar levels in diabetics will lead to increased glucose transport through cell membranes in many cell types (but not apparently in rods due to their high affinity glucose transporters) (130, 131). On entering the tricarboxylic cycle, the excess sugars will lead to an increased demand for oxygen. This also results in the formation of advanced glycosylation end products (AGEs), which are modifications of proteins or lipids that become non-enzymatically glycosylated when exposed to aldose sugars and oxidative stress (213-215). Recent experimental evidence shows that rod photoreceptor cells are major contributors to diabetes-induced oxidative stress by producing reactive oxygen species and local inflammation in the retina (216).
These changes accumulate with time affecting both intracellular and extracellular function and by engaging the receptor for advanced glycation end products (RAGE). Activation of RAGE by AGEs causes upregulation of the transcription factor nuclear factor κ β (NFκβ) and its target genes. In Müller cells this promotes the expression of glial fibrillary acidic protein (GFAP) which is seen very early in diabetes, and the secretion of the cytokine, VEGF. As already alluded to above, VEGF causes changes to small vessels and makes them leaky (VEGF was initially identified as a vascular permeability factor [VPF] (217). Other AGEs contribute to the increase in basal membrane thickness which occurs in many tissues, including the capillaries of the retinal circulation (218-222). All these factors reduce oxygen transport into the retina. Additional biochemical alterations are known to occur in the diabetic retina, but once hypoxia is established, it will cause a cascade of effects that drive the production of VEGF (Figure 16) leading to further changes associated with diabetic retinopathy: and these changes in turn will exacerbate any underlying hypoxia.
Since a major cause of retinal hypoxia is the demand of the rods during periods of dark adaptation, reducing rod dark current should ameliorate diabetic changes. However, many other biochemical changes seen in diabetic retina have been connected to the overproduction of reactive oxygen species. (223). If these pathways are more important than the rod oxygen demand, increasing the retinal oxygen tension could either have little effect, or might even accelerate the progression of diabetic retinopathy. However, pan-retinal laser photocoagulation, the mainstay of treatment for proliferative diabetic retinopathy, has been shown to restore local oxygen levels to normal within weeks of its application (224). It is thought to work mainly by destroying large numbers of rods in the peripheral retina thereby increasing the oxygen supply to the remaining tissue (225). Furthermore, vascular responsiveness to oxygen inhalation also improves following photocoagulation (226-228).
12. Evidence for a new treatment based on the hypothesis that retinal hypoxia can be reduced by keeping rods light adapted at night
The success of panretinal photocoagulation is mainly because the treatment destroys rod photoreceptors, thereby reducing oxygen demand of the outer retina, particularly under dark adapted conditions. The remaining rods benefit, but even more importantly, there is improved ability of oxygen to reach the inner retina from the choroid because of the reduction in the number of rod photoreceptors with their oxygen-absorbing mitochondria. This may change the balance in the supply of oxygen to the inner retina, which already has a compromised retinal circulation.
The use of anti-VEGF drugs is now the most common initial treatment for diabetic eye disease, and hypoxia is the major determinant of VEGF production, but this is almost a circular argument. Direct measurement of oxygen tension in diabetic cat retina shows that the inner retina is more hypoxic than that of normal felines even in regions where there is no capillary dropout evident (229, 230) (Figure 17).
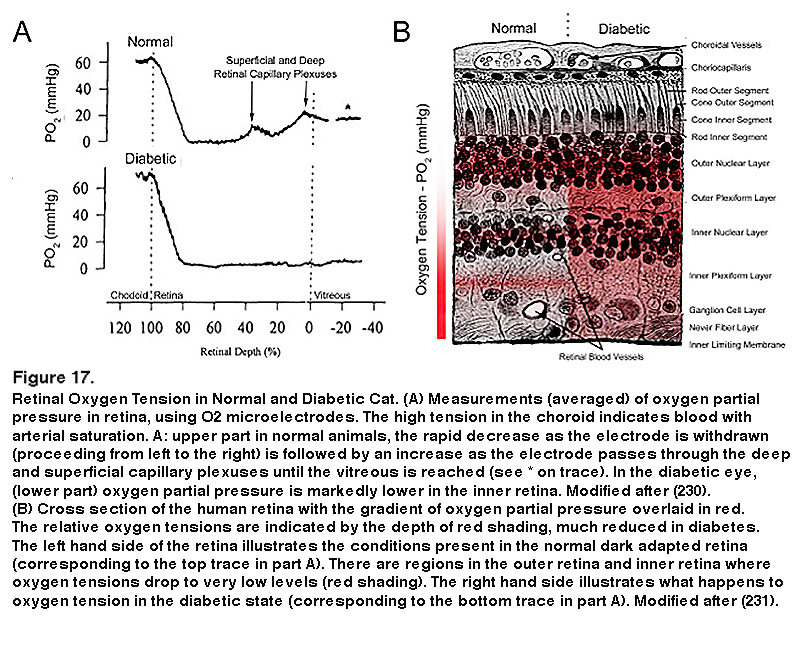
The partial pressure of oxygen falls rapidly from the arterial level when the electrode moves from the RPE into the outer retina, and in darkness reaches a level of zero in the region of the rod inner limb mitochondria (229, 230). This rate of decline is the maximum theoretically possible if the choroid is an infinite source of oxygen, and the mitochondria, an infinite sink. As the electrode tip moves into the inner vascularized retina (see Figure 17A, top left tracing) the partial pressure increases again. Calculation shows that the rod mitochondria must also be absorbing oxygen from the retinal vessels. In light the oxygen tension is higher for all retinal locations, but especially at the level of the rod inner limbs, where it increases by about 30 mm Hg. (not illustrated). This shows how rapid the rate of respiration is, and how quickly it is affected by the processes of transduction. In addition in diabetic cat retina, even in regions in which there is no capillary dropout, the inner retinal oxygen tension is lower than in normal retina (Figure 17). There are also species differences in the oxygen tension profiles. In the miniature pig (224) and macaque (232), although the profiles resemble those shown in Figure 17A (from the cat (211)), the pO2 at the level of the inner limbs does not fall to zero but about 5 mmHg. Nevertheless the outer retinal oxygen consumption greatly exceeds the inner retinal oxygen consumption. In guinea pigs (232) and rabbits (232, 235) animals with poorly developed retinal vessels, more of the inner retina’s oxygen supply is provided by the choroid, but the same general relationships may be seen, and oxygen consumption and hypoxia of the outer retina increases as the photoreceptors develop and their outer limbs lengthen. In the species studied in dark adaptation, diabetic animals have an inner retinal hypoxia more evident than in controls (224, 234). However, in the rat retina, the retinal profiles are different especially in the RCS rat (232), with the minimal pO2 being found in the inner not the outer retina. Nevertheless the difference in oxygen consumption when in light or darkness is greatest in the outer retina, and corresponds to photoreceptor usage. This is supported by the change in profiles in the developing retina, and the change in profile when photoreceptor degenerations develop (233-235). So, in rat (199), direct measurements of intraretinal pO2 using oxygen microelectrodes have not shown hypoxia or change in diabetes. However, this last observation is difficult to reconcile with the other data measured in larger eyes, which are similar to human eye size (195, 235, 236) and to indirect evidences of rod-associated retinal hypoxia (237-239).
Human beings have many inherited diseases that selectively destroy rods, producing retinal dystrophies – Retinitis Pigmentosa is the most common of these conditions. Rod loss in many cases is slow, and diabetes can occur independently in conjunction with the slow loss of photoreceptors. However, in such patients, diabetic retinopathy-type changes rarely happen (Table 1) (240).
Another line of evidence comes from patients with maternally inherited diabetes and deafness (MIDD). These patients have mutation of mitochondrial DNA at position 3243 which manifests in a number of ways and can cause a type of retinal degeneration. This may or may not occur together with diabetes. If the retinal degeneration occurs together with the diabetes, the incidence of microaneurysms is less than if the diabetes occurs alone (241-243).
In some individuals, the efficiency of hypoxia in increasing VEGF and VEGF RNA levels is much reduced and can be seen by incubating white blood cells under normoxic and hypoxic conditions. Among this group, retinopathy does not appear in individuals who also have diabetes (243). In addition, breathing oxygen improves several visual functions in patients with diabetic retinopathy and DME (192, 193, 196, 189, 244, 245).
Finally, patients with diabetes who have sleep apnea, (defined as more than 5 periods, called dips, when oxyhemoglobin saturation falls below 90%), have more rapid development of diabetic eye complications than those who do not have sleep apnea. The increased risk is reduced in patients with sleep apnea who are treated with positive pressure ventilation (246-250). There are several recent reviews that include a more detailed discussion of the hypothesis that rod photoreceptor dark adaptation causes hypoxia and consequent damage to the retina in diabetic retinopathy (251, 252).
Given all the above evidence that hypoxia in the inner retina in diabetic patients causes the vascular damage of diabetic retinopathy, and evidence that rods are responsible for most of the oxygen consumption during their active metabolic time in darkness, it seems reasonable to suggest that curbing the rod activity at night would be beneficial. A simple treatment for impending diabetic retinopathy might be to prevent rod activity during darkness by the application of constant nocturnal light. This is the basis for the new treatment paradigm described below.
We have carried out clinical trials, in which patients wear “light masks” which provide retinal illumination through the closed eyelids during sleep. Sufficient light penetrates the lid to partially light adapt the rods. The light sources were either chemo-luminescent (glow-patches) or light-emitting diodes (LEDs). The green or blue-green light was used to selectively stimulate rods (95% of all photoreceptors are rods) rather than cones, because the sensations aroused by rod stimulation are less intrusive and tend to fade to insignificance as patients adapt to a low background luminance. In order to reduce inter-personal differences, the subjects wore masks which illuminated only one eye, with the other, in darkness being used as a control (253).
The results of a 6 month Phase I trial showed that no adverse reactions were encountered and patients accepted the idea of wearing a mask during sleep. However, unusual for a Phase 1 trial, the therapy caused a significant improvement in the 10 trial eyes relative to their controls. Figure 18 shows the results in one patient.
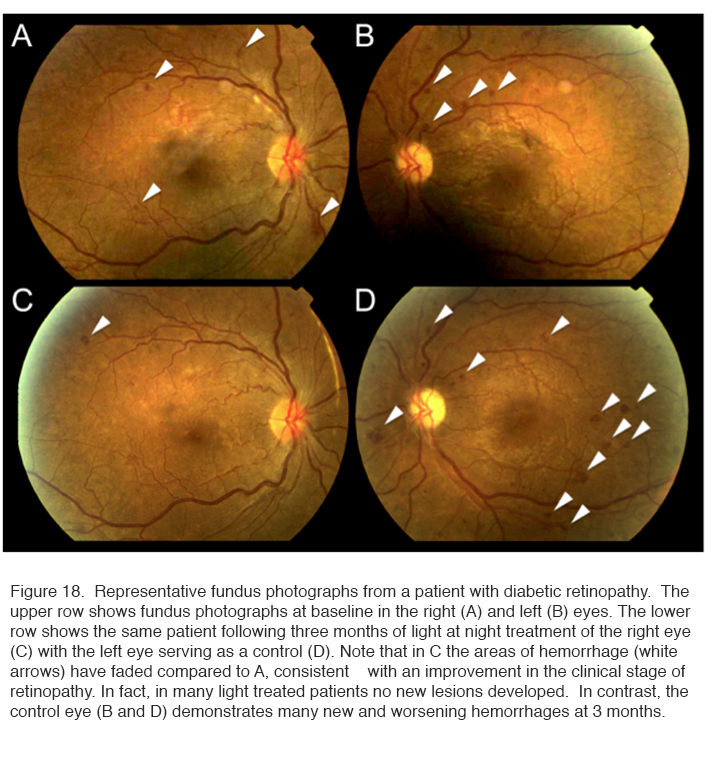
Fundus photographs such as shown in Figure 18 were examined under high magnification and the total areas of microaneurysms measured. The improvement in the condition (decrease in areas of affected central retina) was significant. A similar result was obtained when changes in tritan – blue-yellow – colour vision (known to be damaged early in diabetic retinopathy) was assessed (253) (not shown).
In a Phase II trial of 42 patients with non-center-involving DME, a similar benefit was shown. A high proportion of the treated eyes improved, while this did not occur in untreated eyes (Table 2). Much of this occurred during the first 3 months. Furthermore, whereas four of the patients recruited to the trial dropped out because the unilluminated fellow eyes received laser or intravitreal bevacizumab for DME during the study, none of the illuminated eyes required such treatment.
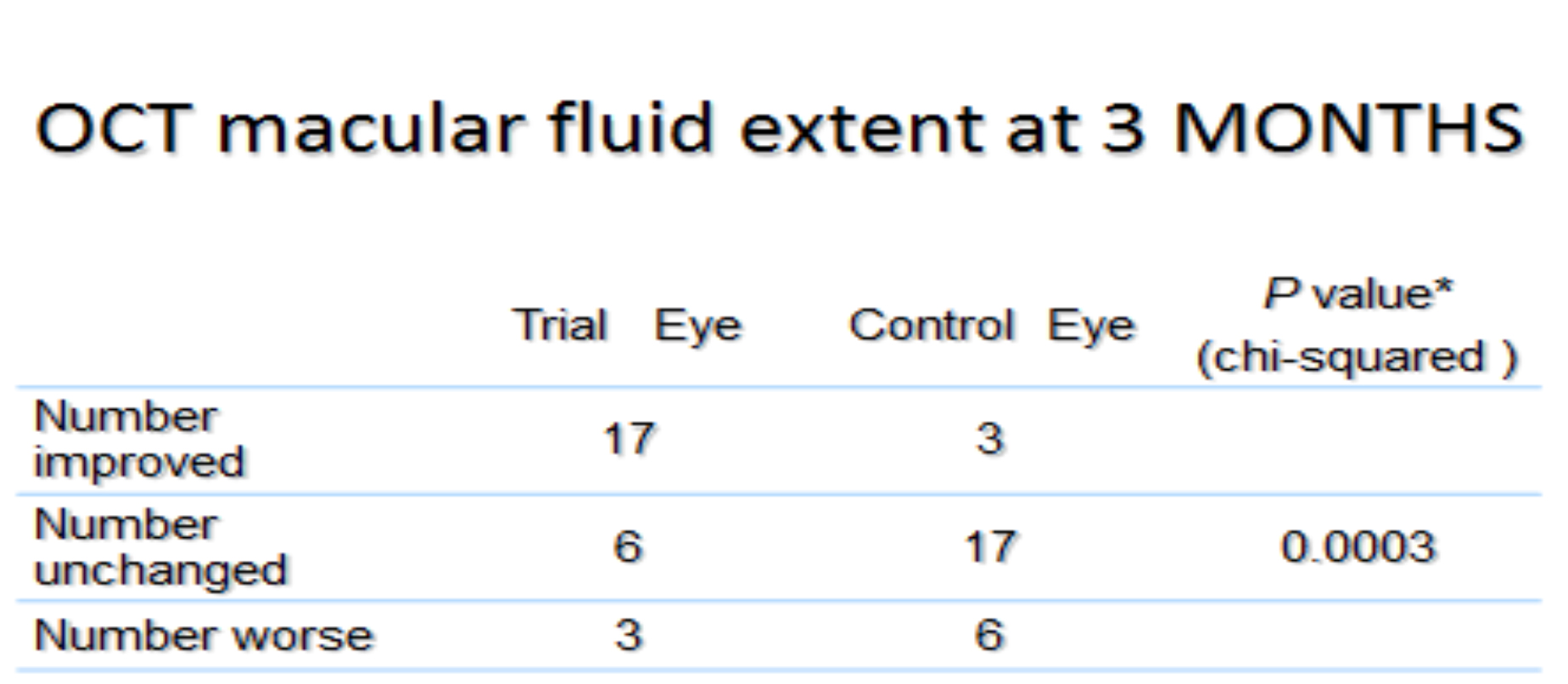
Ocular coherence tomography showed that cysts characteristic of DME disappeared, and significant reduction in thickness occurred (Figure 19) (254). The results of these changes in OCT measurements are shown in Table 3.
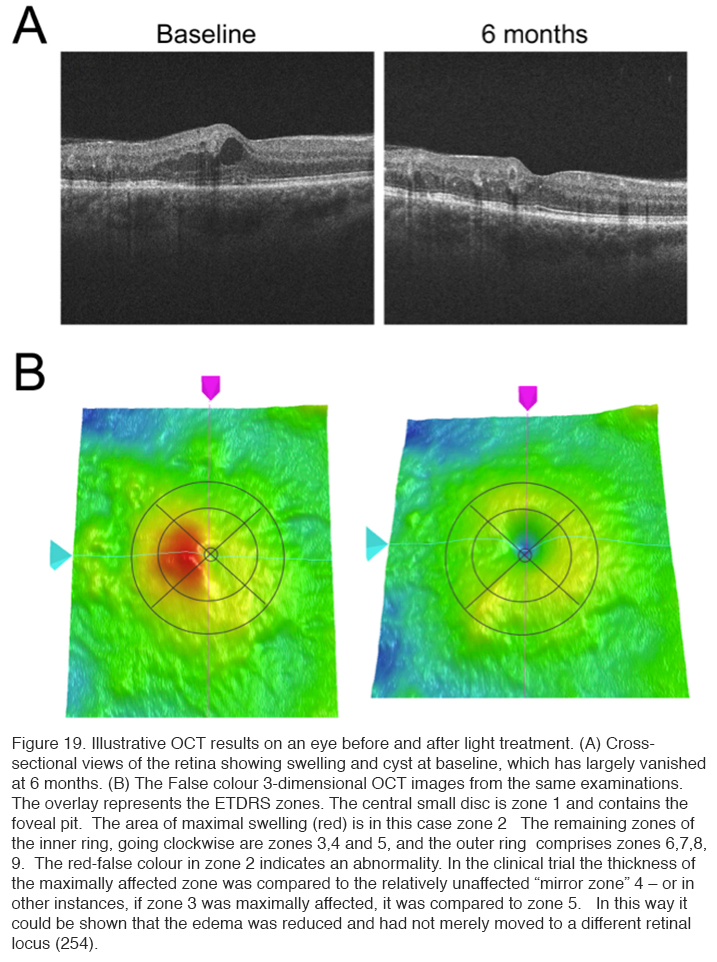

The patients in this trial had very early DME, and in many patients the swelling did not involve the area within 300 μm of the fovea. Hence if there is a reduction observed in local swelling in one retinal region, might swelling simply be occurring in another region? In order to evaluate this, measurements were made in the Early Treatment Diabetic Retinopathy (ETDRS) zones, and the region with maximal swelling was compared to its opposite “mirror” zone. It can be seen that decrease in thickness occurs only in treated eyes (Figure 20).
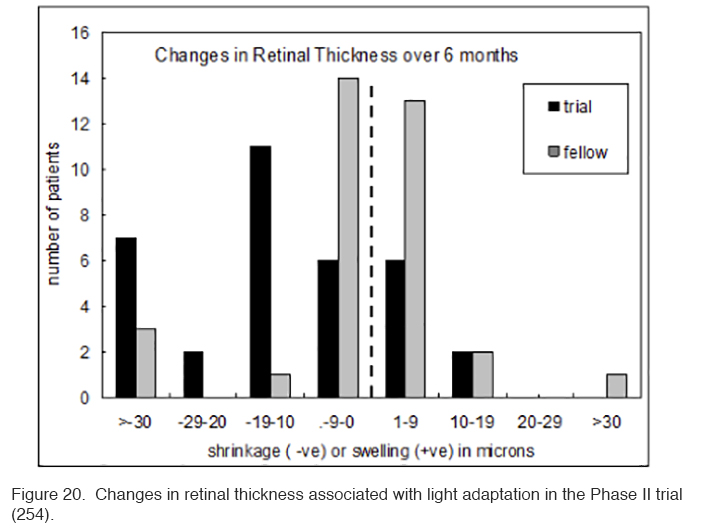
A small but significant improvement in visual acuity and contrast sensitivity also occurred among the light adapted eyes in this trial (table 4). It should be noted, that almost all the patients recruited were at an early stage of diabetic retinopathy with DME and had good visual acuity at baseline so a larger average increase in acuity would not be expected.

Such studies provide a “Proof of Principle” that retinal hypoxia in darkness must be an important driving force for diabetic retinopathy. However, further work is required to establish the role that ‘light-at-night’ may play in the treatment or prevention of the various complications of diabetic eye disease. Furthermore, we do not know how ‘light at night’ interacts with the other treatments for the condition. For this Phase III and IV clinical trials are required. This presents a difficulty, because for new pharmaceuticals, such trials are largely funded directly by the drug companies, while there is no such equivalent for medical devices. Small commercial companies have manufactured masks which can be used in such trials, and apparently in one trial carried out in the Department of Ophthalmology, Vinohrady, Prague, Czech Republic, produced encouraging results. These results have not been published in detail yet. Further trials are starting in London (255) Cardiff (UK) (256) and Boston (USA). There is a considerable need for this research because the cost of the present day “gold standard” treatment of repeated injections is becoming unsustainable (257). (Figure 21 refers of age related macular degeneration, but for diabetes the same economic criteria will apply).
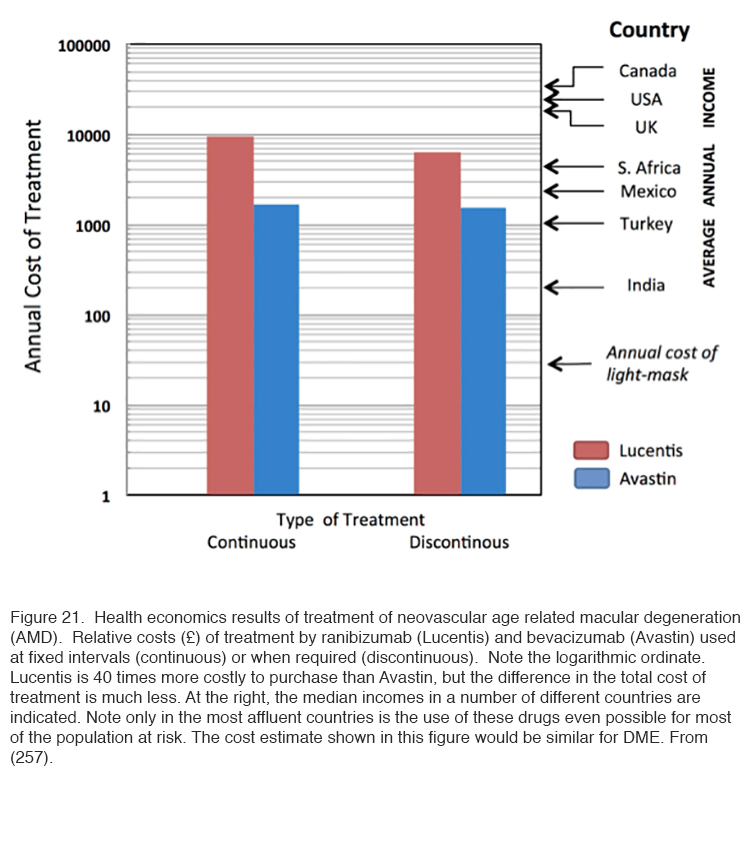
Anti-VEGF drugs are expensive: Avastin costs about 1/40th of the price of Lucentis, but the cost of treatment by the two is not so different, in part because of the associated cost of administration (Figure 21). But only in the first world are incomes high enough for a significant proportion of the population with vision threatening diabetic eye disease to access treatment (257) and even then many individuals with vision threatened by complications from diabetes are not accessing available eye care (258). Worldwide there is not only a shortage of funding for available treatments, but a severe shortage of personnel trained to diagnose the condition and manage it appropriately (259). By contrast, the requirements of light-based treatment to interrupt nocturnal dark adaptation, for example through the use of a light masks, are relatively inexpensive, and so safe and easy to use that it could be made available with only basic instruction. This treatment may not replace medical intervention, but promises to complement or reduce the need for traditional therapies, thereby bringing down the cost of caring for patients with diabetic eye disease. We propose that more research and development be done so that light masks (or a similar device) may be manufactured on a scale that will reduce the cost to a level reaching the masses with diabetic eye disease.
It is possible that the evidence presented above concerning preventing dark adaptation in the diabetic retina, may also apply to other retinal conditions in which anti-VEGF drugs have been shown to be beneficial: Retinal diseases such as “wet” age related macular degeneration (257-265) (see chapter on age related macular degeneration (AMD) in Webvision) and retinopathy of prematurity (266) .
Acknowledgements
The authors thank Carol Spencer (Lahey Hospital Library) for research support. Figures 3, 4 and 5 were kindly provided by the ocular photography department of the Moran Eye Center. Neither of the authors have any direct financial interest in any devices used in the clinical work described.
About the Authors

Geoffrey Arden graduated in 1951 from University College London (UCL) and joined the MRC Vision Research Unit in the newly formed Institute of Ophthalmology. He obtained a Ph.D. in photochemistry under HJA Dartnall, and returned to UCL to complete a medical degree. After studying neurophysiological techniques with Ragnar Granit in Stockholm and KT Brown in San Francisco, he returned to the Institute of Ophthalmology. In the absence of any equipment except an ECG he devised the electro-oculogram (EOG) as a clinical test and developed the first clinical electrophysiological department in the UK. He was appointed Professor, Honorary Consultant at Moorfields Eye Hospital and Director of the Electrodiagnostic department in 1968. He pioneered the use of cortical evoked potentials and the pattern ERG and devised clinical techniques for the measurement of achromatic and colour-contrast sensitivity. In an emeritus capacity he continues projects with investigators from Europe and the US and has turned his attention to the causes and treatment of diabetic retinopathy. He served a term as Chief Editor of the journal Vision Research.
geoffreyarden@aol.com

Dr. David J. Ramsey received his M.D. and Ph.D. degrees from the University of Illinois at Chicago in 2008. He completed his residency in Ophthalmology at the Wilmer Eye Institute of the Johns Hopkins School of Medicine in 2012 followed by a combined medical/surgery fellowship in vitreoretinal diseases at the Massachusetts Eye and Ear Infirmary in 2014. Dr. Ramsey is currently a senior staff physician at the Lahey Clinic and sees patients in Burlington, MA. He also sees patients and performs surgery in Peabody, MA. His research focuses on the prevention of dark adaptation in patients with diabetes as a means to ameliorate diabetic changes in the retina. Future clinical trials will seek to determine whether this noninvasive, environmental intervention can limit the progression of diabetic retinopathy to PDR and/or reduce levels of sight-impairing macular edema.
ramsey@post.harvard.edu
References
1: Health Topics, Noncommunicable Diseases, Diabetes. Regional Office for Europe, World Health Organization.
2: National Diabetes Statistics Report, 2014. National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention.
3: Facts About Diabetic Retinopathy. National Eye Institute.
4: Liew G1, Michaelides M, Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16-64 years), 1999-2000 with 2009-2010. BMJ Open. 2014 ;4:e004015. [PubMed]
5: Adams, F in Adams, F ed (1856): The extant works of Aretaeus the Cappadocian, p. 338. London: The Sydenham Society. [PubMed]
6: Allen FM. Current judgments on metabolic control and complications in diabetes. N Engl J Med. 1953; 248: 133-136. [PubMed]
7: Ramsey DJ, Sunness JS. “Pregnancy and the Mother’s Eye.” In Tasman W., Jaeger E.A., eds., Duane’s Clinical Ophthalmology, Vol. 5, Chapter 32, 2013.
8: Vaxillaire M, Froguel P. Monogenic diabetes in the young, pharmacogenetics and relevance to multifactorial forms of type 2 diabetes. Endocr Rev. 2008; 29: 254-64. doi: 10.1210. [PubMed]
9: Abdul-Rasoul M, Habib H, Al-Khouly M. ‘The honeymoon phase’ in children with type 1 diabetes mellitus: frequency, duration, and influential factors. Pediatr Diabetes. 2006; 7: 101-7. [PubMed]
10: Daneman D. Type I Diabetes. Lancet. 2006; 9513: 847-858. [PubMed]
11: Kelly MA, Mijovic CH, Barnett AH. Genetics of type 1 diabetes. Best Pract Res Clin Endocrinol Metab. 2001; 15: 279-91. [PubMed]
12: Dabelea D, Mayer-Davis EJ, Saydah S, Imperatore G, Linder B, Divers J, Bell R, Badaru A, Talton JW, Crume T, Liese AD, Merchant AT, Lawrence JM, Reynolds K, Dolan L, Liu LL, Hamman RF; SEARCH for Diabetes in Youth Study. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014; 311: 1778-86. [PubMed]
13: Dabelea D, Ma Y, Knowler WC, Marcovina S, Saudek CD, Arakaki R, White NH, Kahn SE, Orchard TJ, Goldberg R, Palmer J, Hamman RF; Diabetes Prevention Program Research Group. Diabetes autoantibodies do not predict progression to diabetes in adults: Diabet Med. 2014; 31: 1064-8. [PubMed]
14: Taplin CE, Craig ME, Lloyd M, Taylor C, Crock P, Silink M, Howard NJ. The rising incidence of childhood type 1 diabetes in New South Wales, 1990-2002. Med J Aust. 2005; 183: 243-6. {PubMed]
15: Hyöty H, Hiltunen M, Knip M, Laakkonen M, Vähäsalo P, Karjalainen J, Koskela P, Roivainen M, Leinikki P, Hovi T, et al A. prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes. 1995; 44: 652-7. [PubMed]
16: Zhao M, Pan SS. Discussion of correlative factors and risk in patients with diabetic erectile dysfunction. Zhonghua Nan Ke Xue. 2002; 8: 395-7. Chinese. [PubMed]
17: Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for disease control and prevention 2011.
18: Leung MY, Pollack LM, Colditz GA, Chang SH. Life years lost and lifetime health care expenditures associated with diabetes in the U.S., National Health Interview Survey, 1997-2000. Diabetes Care. 2015; 38: 460-8. doi: 10.233. [PubMed]
19: Lueder GT, Silverstein J; American Academy of Pediatrics Section on Ophthalmology and Section on Endocrinology. Screening for retinopathy in the pediatric patient with type 1 diabetes mellitus. Pediatrics. 2005; 116: 270-3. [PubMed]
20: Verougstraete C, Toussaint D, De Schepper J, Haentjens M, Dorchy H. First microangiographic abnormalities in childhood diabetes–types of lesions. Graefes Arch Clin Exp Ophthalmol. 1991; 229: 24-32. [PubMed]
21: Younis N, Broadbent DM, Harding SP, Vora JP. Incidence of sight-threatening retinopathy in Type 1 diabetes in a systematic screening programme. Diabet Med. 2003; 20: 758-65. [PubMed]
22: Eastman RC, Javitt JC, Herman WH, Dasbach EJ, Zbrozek AS, Dong F, Manninen D, Garfield SA, Copley-Merriman C, Maier W, Eastman JF, Kotsanos J, Cowie CC, Harris M. Model of complications of NIDDM. I. Model construction and assumptions. Diabetes Care. 1997; 20: 725-34. [PubMed]
23: Malone JI, Morrison AD, Pavan PR, Cuthbertson DD; Diabetic Control and Complications Trial. Prevalence and significance of retinopathy in subjects with type 1 diabetes of less than 5 years’ duration screened for the diabetes control and complications trial. Diabetes Care. 2001; 24: 522-6. [PubMed]
24: Klein R, Meuer SM, Moss SE, Klein BE. Retinal microaneurysm counts and 10-year progression of diabetic retinopathy. Arch Ophthalmol. 1995; 113: 1386-91. [PubMed]
25: [No authors listed] Epidemiology of Diabetes Interventions and Complications (EDIC). Design, implementation, and preliminary results of a long-term follow-up of the Diabetes Control and Complications Trial cohort. Diabetes Care. 1999; 22: 99-111. [PubMed]
26: Younis N, Broadbent DM, Vora JP, Harding SP; Liverpool Diabetic Eye Study. Incidence of sight-threatening retinopathy in patients with type 2 diabetes in the Liverpool Diabetic Eye Study: a cohort study. Lancet. 2003; 361(9353): 195-200. [PubMed]
27: Joussen AM Gardner TW Kirchoff B and Ryan SJ ( eds) Retinal Vascular disease. Springer. 2007; p. 778.
28: Henson DB, North RV. Dark adaptation in diabetes mellitus. Br J Ophthalmol. 1979; 63: 539-41. [PubMed]
29: Schulze DP, Freese R, Sauerwald R. Nyktometric examinations on eye function in diabetic retinopathyKlin Monbl Augenheilkd. 1976; 169: 369-72. [PubMed]
30: Abraham FA1, Haimovitz J, Berezin M. The photopic and scotopic visual thresholds in diabetics without diabetic retinopathy. Metab Pediatr Syst Ophthalmol. 1988; 11: 76-7. [PubMed]
31: Greenstein VC, Thomas SR, Blaustein H, Koenig K, Carr RE.Effects of early diabetic retinopathy on rod system sensitivity.. Optom Vis Sci. 1993; 70: 18-23. [PubMed]
32: Birch J, Dain SJ. An averaging method for the interpretation of the Farnsworth-Munsell 100-Hue Test–II. Colour vision defects acquired in diabetic retinopathy. Ophthalmic Physiol Opt. 1987; 7: 281-91. [PubMed]
33: Arden G, Gündüz K, Perry S. Color vision testing with a computer graphics system: preliminary results Doc Ophthalmol. 1988; 69: 167-74. [PubMed]
34::Fong DS, Barton FB, Bresnick GH. Impaired color vision associated with diabetic retinopathy: Early Treatment Diabetic Retinopathy Study Report No. 15. Am J Ophthalmol. 1999; 128: 612-7. [PubMed]
35: Rasmussen ML, Broe R, Feydkjaer-Olson U Olsen BS Mortensen HB, Peto T, Grauslund J. Comparison between Early Treatment Diabetic Retinopathy Study 7-field retinal photos and non-mydriatic, mydriatic and mydriatic steered widefield scanning laser ophthalmoscopy for assessment of diabetic retinopathy. J Diabetes Complications. 2015; 29: 99-104. [PubMed]
36: Silva PS Cavallerano JD, Sun JK, Soliman AZ, Aiello LM, Aiello LP. Peripheral lesions identified by mydriatic ultrawide field imaging: distribution and potential impact on diabetic retinopathy severity. Ophthalmology. 2013; 120: 2587-95. [PubMed]
37: Papachristodoulou D, Heath H, Kang SS. The development of retinopathy in sucrose – sucrose-fed and streptozotocin-diabetic rats. Diabetologia. 1976; 12: 367-74.
38: Kern TS, Engerman RL. Comparison of retinal lesions in alloxan- diabetic rats and galactose-fed rats. Curr Eye Res. 1994; 13: 863-7. [PubMed]
39: Lutty GA. Effects of diabetes on the eye. Invest Ophthalmol Vis Sci. 2013; 54: 13-12979. [PubMed]
40: Wu M, Chen Y, Wilson K, Chirindel A, Ihnat MA, Yu Y, Boulton ME, Szweda LI, Ma JX, Lyons TJ. Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008; 49: 2679-85. [PubMed]
41: Osaadon P, Fagan XJ, Lifshitz T, Levy J. A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye (Lond). 2014; 28: 510-20. [PubMed]
42: Albert and Jakobiec, 1994 Albert & Jakobiec’s Principles & Practice of Ophthalmology: 4-Volume Set and Website R Twerk &Co.
43: [No authors listed] Techniques for scatter and local photocoagulation treatment of diabetic retinopathy: Early Treatment Diabetic Retinopathy Study Report no. 3. The Early Treatment Diabetic Retinopathy Study Research Group. Int Ophthalmol Clin. 1987; 27: 254-64. [PubMed]
44: ETDRS report no. 19. Early Treatment Diabetic Retinopathy Study Research Group. Arch Ophthalmol. 1995; 113: 1144-55.
45: Nordwall M, Bojestig M, Arnqvist HJ, Ludvigsson J; Linköping Diabetes Complications Study. Declining incidence of severe retinopathy and persisting decrease of nephropathy in an unselected population of Type 1 diabetes-the Linköping Diabetes Complications Study. Diabetologia. 2004;47: 1266-72. Epub 2004 Jul 2. [PubMed]
46: [No authors listed] Focal photocoagulation treatment of diabetic macular edema. Relationship of treatment effect to fluorescein angiographic and other retinal characteristics at baseline: ETDRS report no. 19. Early Treatment Diabetic Retinopathy Study Research Group. Arch Ophthalmol. 1995; 113: 1144-55.
47: Blumenkranz MS. Optimal current and future treatments for diabetic macular oedema. Eye (Lond). 2010; 24: 428-34. [PubMed]
48: Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol. 1984; 102: 527-32. [PubMed]
49: Klein R., Klein B.E.K., Moss S.E. The Wisconsin epidemiologic study of diabetic retinopathy. XV. The long term incidence of macular edema. Ophthalmology. 1995; 102: 7–16. [PubMed]
50: [No authors listed] Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group Arch Ophthalmol. 1985; 103: 1796-806. [PubMed]
51: [No authors listed] Fluorescein angiographic risk factors for progression of diabetic retinopathy. ETDRS report number 13. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology. 1991; 98 Suppl: 834-40. [PubMed]
52: Bresnick GH, De Venecia G, Myers FL, Harris JA, Davis MD. Retinal ischemia in diabetic retinopathy. Arch Ophthalmol. 1975; 93: 1300-10. [PubMed]
53: Panozzo G, Gusson E, Parolini B, Mercanti A. Role of OCT in the diagnosis and follow up of diabetic macular edema. Semin Ophthalmol. 2003; 18: 74-81. [PubMed]
54: Brown JC, Solomon SD, Bressler SB, Schachat AP, DiBernardo C, Bressler NM. Detection of diabetic foveal edema: contact lens biomicroscopy compared with optical coherence tomography. Arch Ophthalmol. 2004; 122: 330-5. [PubMed]
55: Wu L, Fernandez-Loaiza P, Sauma J, Hernandez-Bogantes E, and Masis M Classification of diabetic retinopathy and diabetic macular edema. World J Diabetes. 2013; 4: 290–294. [PubMed]
56: Diabetic retinopathy study. Report Number 6. Design, methods, and baseline results. Report Number 7. A modification of the Airlie House classification of diabetic retinopathy. Prepared by the Diabetic Retinopathy. Invest Ophthalmol Vis Sci. 1981; 21(1 Pt 2): 1-226.
57: Early Treatment Diabetic Retinopathy Study Research Group. Grading diabetic retinopathy from stereoscopic color fundus photographs–an extension of the modified Airlie House classification. ETDRS report number 10. Ophthalmology.1991; 98(5 Suppl): 786-806.
58: Haut J, Rousselie F, Guyot C, Pinon F, Dureuil J, Baltazis S. Treatment of diabetic retinopathy by centripetal pan-retinal photocoagulation with the argon laser Arch Ophtalmol Rev Gen Ophtalmol. 1975; 35: 951-60. [PubMed]
59: Coscas G, Chaine G. Treatment of diabetic retinopathy with laser photocoagulation. Diabetes Metab. 1979; 5: 247-59. [PubMed]
60: Jyothi S, Sivaprasad S. Five-year visual outcome following laser photocoagulation of diabetic macular. Eye (Lond). 2011; 25: 851-8. [PubMed]
61: Lim MC, Tanimoto SA, Furlani BA, Lum B, Pinto LM, Eliason D, Prata TS, Brandt JD, Morse LS, Park SS, Melo LA Jr. Effect of diabetic retinopathy and panretinal photocoagulation on retinal nerve fiber layer and optic nerve appearance. Arch Ophthalmol. 2009; 127: 857-62. [PubMed]
62: [No authors listed] Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Arch Ophthalmol. 1985; 103: 1796-806. [PubMed]
63: Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, Gibson A, Sy J, Rundle AC, Hopkins JJ, Rubio RG, Ehrlich JS; RISE and RIDE Research Group. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology. 2012; 119: 789-801. [PubMed]
64: Sivaprasad S, Elagouz M, McHugh D, Shona O, Dorin G. Micropulsed diode laser therapy: evolution and clinical applications. Surv Ophthalmol. 2010; 55: 16-30. [PubMed]
65: Friberg TR, Karatza EC. The treatment of macular disease using a micropulsed and continuous wave 810-nm diode laser. Ophthalmology. 1997; 104: 2030-8. [PubMed]
66: Figueira J1, Khan J, Nunes S, Sivaprasad S, Rosa A, de Abreu JF, Cunha-Vaz JG, Chong NV. Prospective randomised controlled trial comparing sub-threshold micropulse diode laser photocoagulation and conventional green laser for clinically significant diabetic macular oedema. Br J Ophthalmol. 2009; 93: 1341-4. [PubMed]
67: Stone J1, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995; 15: 4738-47. [PubMed]
68: Provis JM1, Leech J, Diaz CM, Penfold PL, Stone J, Keshet E . Development of the human retinal vasculature: cellular relations and VEGF expression. Exp Eye Res. 1997; 65: 555-68. [PubMed]
69: Murata T1, Ishibashi T, Khalil A, Hata Y, Yoshikawa H, Inomata H. Vascular endothelial growth factor plays a role in hyperpermeability of diabetic retinal vessels. Ophthalmic Res. 1995; 27: 48-52. [PubMed]
70: Murata T1, Nakagawa K, Khalil A, Ishibashi T, Inomata H, Sueishi K. The relation between expression of vascular endothelial growth factor and breakdown of the blood-retinal barrier in diabetic rat retinas. Lab Invest. 1996; 74: 819-25. [PubMed]
71: Campochiaro PA, Wykoff CC, Shapiro H, Rubio RG, Ehrlich JS. Neutralization of vascular endothelial growth factor slows progression of retinal nonperfusion in patients with diabetic macular edema. Ophthalmology. 2014; 121: 1783-9. [PubMed]
72: Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994; 331: 1480-7. [PubMed]
73: Rosenfeld PJ, Schwartz SD, Blumenkranz MS, Miller JW, Haller JA, Reimann JD, Greene WL, Shams N. Maximum tolerated dose of a humanized anti-vascular endothelial growth factor antibody fragment for treating neovascular age-related macular degeneration. Ophthalmology. 2005; 112:1048-53. [PubMed]
74: Andreoli, C. M. & Miller, J. W. Anti-vascular endothelial growth factor therapy for ocular neovascular disease. Current opinion in ophthalmology 2007; 18: 502-508. [PubMed]
75: Chen HX1, Gore-Langton RE, Cheson BD. Clinical trials referral resource: Current clinical trials of the anti-VEGF monoclonal antibody bevacizumab. Oncology (Williston Park). 2001; 15: 1017.
76: Rosenfeld, P. J., Moshfeghi, A. A. & Puliafito, C. A. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmic surgery, lasers & imaging. 2005; 36: 331-335. [PubMed]
77: Nicholson, B. P. & Schachat, A. P. A review of clinical trials of anti-VEGF agents for diabetic retinopathy. V. Graefe’s Arch. Clin exper. Ophthalmol 2010; 248: 915-930. [PubMed]
78: Stefanini FR, Badaro E, Falabella P, Koss M, Farah ME and Maia M. Anti-VEGF for the management of Diabetic Macular Edema . J. Immunolog Res 2014 article ID 632307. [PubMed]
79: Dafer RM, Schneck M, Friberg TR, Jay WM. Intravitreal ranibizumab and bevacizumab: a review of risk. Semin Ophthalmol. 2007; 22: 201-4. [PubMed]
80: Azanza Perea JR, García Layana A. Ranibizumab versus bevacizumab. Pharmacological considerations. Arch Soc Esp Oftalmol. 2012 ;87 Suppl 1:3-9. [PubMed]
81: Mitchell P, Bandello F, Schmidt-Erfurth U, Lang GE, Massin P, Schlingemann RO, Sutter F, Simader C, Burian G, Gerstner O, Weichselberger A; RESTORE study group. The RESTORE study: ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology. 2011; 118: 615-25. [PubMed]
82: Salam A, DaCosta J, Sivaprasad S. Anti-vascular endothelial growth factor agents for diabetic maculopathy. Br J Ophthalmol 2010; 94: 821-826. [PubMed]
83: Elman MJ, Aiello LP, Beck RW, Bressler N M , Bressler SB, Edwards AR, Diabetic Retinopathy Clinical Research Network et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular oedema. Ophthalmology 2010; 117: 1064-1077. [PubMed]
84: Michaelides M , Kaines A, Hamilton RD, Fraser-Bell S Rajen am R, Quhi U et al. A prospective randomized trial of intravitreal bevacizumab or laser therapy in the management of diabetic macular oedema (BOLT study) 12-month data: report 2. Ophthalmology 2010; 117: 1078-1086. [PubMed]
85: Azza El-Remessy M , A l i Behzadian A, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol 2003; 162: 1995-2004. [PubMed]
86: Stein JD1, Newman-Casey PA, Kim DD, Nwanyanwu KH, Johnson MW, Hutton DW. Cost-effectiveness of various interventions for newly diagnosed diabetic macular edema. Ophthalmology. 2013; 120: 1835-42. [PubMed]
87: El Sanharawi M, Kowalczuk L, Touchard E, Omri S, de Kozak Y, Behar-Cohen F. Protein delivery for retinal diseases: from basic considerations to clinical applications. Prog Retin Eye Res. 2010; 29: 443-65. [PubMed]
88: Silva PS, Sun JK, Aiello LP. Role of steroids in the management of diabetic macular edema and proliferative diabetic retinopathy. Semin Ophthalmol. 2009; 24: 93-9. [PubMed]
89: Jeon S, Lee WK. Effect of intravitreal triamcinolone in diabetic macular edema unresponsive to intravitreal bevacizumab. Retina. 2014; 341: 606-11. [PubMed]
90: Thomas BJ, Shienbaum G, Boyer DS, Flynn HW Jr. Evolving strategies in the management of diabetic macular edema: clinical trials and current management. Can J Ophthalmol. 2013; 48: 22-30. [PubMed]
91: Yilmaz T, Cordero-Coma M, Lavaque AJ, Gallagher MJ, Padula WV. Triamcinolone and intraocular sustained-release delivery systems in diabetic retinopathy. Curr Pharm Biotechnol. 2011; 12: 337-46. [PubMed]
92: Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805-11. [PubMed]
93: Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006; 47: 1594-9. [PubMed]
94: Kowluru RA, Kowluru V, Xiong Y, Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006 15; 41: 1191-6. [PubMed]
95: Kobayashi T, Oku H, Komori A, Okuno T, Kojima S, Obayashi H, Sugiyama T, Hasegawa G, Fukui M, Nakamura N, Ikeda T: Advanced glycation end products induce death of retinal neurons via activation of nitric oxide synthase. Exp Eye Res 2005; 81: 647–654. [PubMed]
96: Stitt AW, Bhaduri T, McMullen CB, Gardiner TA, Archer DB. Advanced glycation end products induce blood-retinal barrier dysfunction in normoglycemic rats. Mol Cell Biol Res Commun. 2000; 3: 380-8. [PubMed]
97: Pryor WA . OXY-RADICALS AND RELATED SPECIES: Their Formation, Lifetimes, and Reactions. Ann. Rev. Physiol. 1986; 48: 657—67. [PubMed]
98: Grivenkova VG Vinogradov AD. Generation of superoxide by the mitochondrial Complex I. Biochem Biophys Acta 2006; 1757: 553-61.
99: Baines JW. Role of Oxidative Stress in Development of Complications in Diabetes. Diabetes 1991; 40: 405-12. [PubMed]
100: Wu Y, Tang L, Chen B: Oxidative stress: implications for the development of diabetic retinopathy and antioxidant therapeutic perspectives. Oxid Med Cell Longev; 2014; 2014: Article ID 752387. [PubMed]
101: Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006; 8: 691-72. [PubMed]
102: Brownlee M. Biochemistry and molecular Cell Biology of diabetic complications. Nature 2001; 414: 813-820. [PubMed]
103: Lee MY, Griendling KK. Redox signaling, vascular function, and hypertension. Antioxid Redox Signal 2008; 10: 1045-10598. [PubMed]
104: Gupte SA, Wolin MS. Oxidant and redox signaling in vascular oxygen sensing: implications for systemic and pulmonary hypertension. Antioxid Redox Signal 2008; 10: 1137-1152. [PubMed]
105: Kaiser N, Sasson S, Feener EP, et al. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 1993; 42: 80–89. [PubMed]
106: Souza HP, Liu X, Samouilov A, Kuppusamy P, Laurindo FR, Zweier JL. . Quantitation of superoxide generation and substrate utilization by vascular NAD(P)H oxidase. Am J Physiol Heart Circ Physiol. 2002; 282: H466-74. [PubMed]
107: Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008; 134: 279–290. [PubMed]
108: Brownlee M. Biochemistry and molecular Cell Biology of diabetic complications Nature 2001; 414: 813-820 [PubMed]
109: Hancock JT, R. Desikan R, Neill SJ. Role of Reactive Oxygen Species in Cell Signaling Pathways. Biochemical and Biomedical Aspects of Oxidative Modification. Biochemical Society Transactions. 2001; 29: 345-350.
110: Bokoch GM, Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002; 100: 2692-6. [PubMed]
111: Yang W, Block ER. Effect of hypoxia and reoxygenation on the formation and release of reactive oxygen species by porcine pulmonary artery endothelial cells. J Cell Physiol. 1995; 164: 414-23. [PubMed]
112: Cervellati F1, Cervellati C, Romani A, Cremonini E, Sticozzi C, Belmonte G, Pessina F, Valacchi G. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free Radic Res 2014; 48: 303-12. [PubMed]
113: Ng CF, Schafer FQ, Buettner GR, Rodgers VG. The rate of cellular hydrogen peroxide removal shows dependency on GSH: mathematical insight into in vivo H2O2 and GPx concentrations. Free Radic Res. 2007; 41: 201-11. [PubMed]
114: Madsen-Bouterse SA Zhong Q Mohamed G Ho Y-S and Kowluru RA . Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic Res. 2010; 44: 313–321. [PubMed]
115: Kern TS. Interrelationships between the Retinal Neuroglia and Vasculature in Diabetes. Diabetes Metab J. 2014; 38: 163-70 [PubMed]
116: Du Y, Veenstra A, Palczewski K, Kern TS. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci USA. 2013; 110: 16586-91. [PubMed]
117: Kurtenbach A, Mayser HM, Jagle H, Fritsche A , Zrenner E, Hyperoxia , hyperglycaemia and photoreceptor sensitivity in normal and diabetic subjects. Vis. Neurosci 2006; 23: 651-661. [PubMed]
118: Tzekov, R. and Arden, G. B. The electroretinogram in diabetic retinopathy. Surv Ophthalmol. 1999; 44: 53-60.
119: Holfort SK1, Jackson GR, Larsen M. Dark adaptation during transient hyperglycemia in type 2 diabetes. Exp Eye Res. 2010; 91: 710-4. [PubMed]
120: McFarland RA, Forbes WH. The effects of variations in the concentration of oxygen and of glucose on dark adaptation. J Gen Physiol. 1940; 24: 69-98. [PubMed]
121: Holopigian K Greenstein VC Seiple W Hood DC Carr RE . Evidence for Photoreceptor Changes in Patients With Diabetic Retinopathy. Invest Ophthalmol Vis Sci. 1997; 38: 2355-2365. [PubMed]
122: Wong TY, Liew G, Tapp RJ, Schmidt MI, Wang JJ, Mitchell P, Klein R, Klein BE, Zimmet P, Shaw J. Relation between fasting glucose and retinopathy for diagnosis of diabetes: three population-based cross-sectional. Lancet. 2008; 371: 736-43. [PubMed]
123: Kundu D, Mandal T, Nandi M, Osta M, Bandyopadhyay U, Ray D. Oxidative stress in diabetic patients with retinopathy. Ann Afr Med. 2014; 13: 41-6. [PubMed]
124: Tapp RJ, Zimmet PZ, Harper CA, de Courten MP, McCarty DJ, Balkau B, Taylor HR, Welborn TA, Shaw JE. Aus Diab Study Group. Diagnostic thresholds for diabetes: the association of retinopathy and albuminuria with glycaemia. Diabetes Res Clin Pract. 2006; 73: 315-21. [PubMed]
125: Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest Ophthalmol Vis Sci. 2011; 52: 8739-46. [PubMed]
126: Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805-11. [PubMed]
127: Kolm-Litty V, Sauer U, Nerlich A, Lehmann and Schleicher ED . High Glucose induced Transforming Growth Factor β1 Production is mediated by the hexosamine Pathway in porcine Glomerular Mesangial cells. J Clin Invest. 1998; 101: 160-9. [PubMed]
128: Small M, Alzaid A, MacCuish AC. Diabetic hyperosmolar non-ketotic decompensation. J Med. 1988; 66: 251-7. [PubMed]
129: Ceriello A, Esposito K, Piconi L, et al. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008; 57: 1349-54. [PubMed]
130: Busik JV, Olson LK, Grant MB, Henry DN. Glucose-induced activation of glucose uptake in cells from the inner and outer blood–retinal barrier. Invest Ophthalmol Vis Sci. 2002; 43: 2356-63. [PubMed]
131: Busik JV, Mohr S, Grant MB. Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008; 57: 1952-65. [PubMed]
132: Kador PF, Akagi Y, Terubayashi H, Wyman M, Kinoshita JH. Prevention of pericyte ghost formation in retinal capillaries of galactose-fed dogs by aldose reductase inhibitors. Archives of Ophthalmology. 1988; 106: 1099-1102. [PubMed]
133: Gabbay KH. The sorbitol pathway and the complications of diabetes. The New England Journal of Medicine. 1973; 288: 831–836: [Pubmed]
134: Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998; 47: 859–866. [PubMed]
135: Aiello LP, Bursell SE, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective β-isoform-selective inhibitor. Diabetes. 1997; 46: 1473. [PubMed]
136: Tarr JM, Kaul K, Chopra M, Kohner EM, Chibber R. Pathophysiology of diabetic retinopathy. ISRN Ophthalmol. 2013; 2013: 343560. [PubMed]
137: Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007; 2007: 43603. [PubMed]
138: Zhang X, Zeng H, Sheian B, Wang N, Gillies MC. Diabetic macular edema: new concepts in patho-physiology and treatment. Cell & Bioscience. 2014; 4: 27-41. [PubMed]
139: Yang W, Block ER. Effect of hypoxia and reoxygenation on the formation and release of reactive oxygen species by porcine pulmonary artery endothelial cells. J Cell Physiol. 1995; 164: 414-23. [PubMed]
140: Bernardini A, Brockmeier U, Metzen E, Berchner-Pfannschmidt U, Harde E, Acker-Palmer A, Papkovsky D, Acker H, Fandrey J. Type I cell ROS kinetics under hypoxia in the intact mouse carotid body ex vivo: a FRET based study. Am J Physiol Cell Physiol. 2015; 308.1: C61-C67. [PubMed]
141: Eshaq RS, Wright WS, Harris NR. Oxygen delivery, consumption, and conversion to reactive oxygen species in experimental models of diabetic retinopathy. Redox Biol. 2014; 2: 661-6. [PubMed]
142: Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol 1985. 2000; 88: 1880-9. [PubMed]
143: Chandel NS1, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000; 275: 25130-8. [PubMed]
144: Strasser A, Stanimirovic D, Kawai N, McCarron RM, Spatz M. Hypoxia modulates free radical formation in brain microvascular endothelium. Acta Neurochir Suppl. 1997; 70: 8-11. [PubMed]
145: Zuo L, Shiah A, Roberts WJ, Chien MT, Wagner PD, Hogan MC. Low PO₂ conditions induce reactive oxygen species formation during contractions in single skeletal muscle fibers. Am J Physiol Regul Integr Comp Physiol. 2013; 304: R1009-16. [PubMed]
146: Hernansanz-Agustín P, Izquierdo-Álvarez A, Sánchez-Gómez FJ, Ramos E, Villa-Piña T, Lamas S, Bogdanova A, Martínez-Ruiz A. Acute hypoxia produces a superoxide burst in cells. Free Radic Biol Med. 2014; 71: 146-56. [PubMed]
147: Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res. 2009; 82: 9-20. [PubMed]
148: Jakus, V Rietbock N. Advanced Glycation End-Products and the Progress of Diabetic Vascular Complications. Physiol. Res. 2004; 53: 131-142. [PubMed]
149: J W. Baynes and S R. Thorpe. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999; 48: 1–9. [PubMed]
150: Yamada H1, Yamada E, Hackett SF, Ozaki H, Okamoto N, Campochiaro PA. Hyperoxia causes decreased expression of vascular endothelial growth factor and endothelial cell apoptosis in adult retina. J Cell Physiol. 1999; 179: 149-56. [PubMed]
151: Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805-11. [PubMed]
152: Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006; 47: 1594-9. [PubMed]
153: Kowluru RA, Kowluru V, Xiong Y, Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006; 41: 1191-6. [PubMed]
154: Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 2000; 275: 16023-16029. [PubMed]
155: Mishra M and Kowluru RA. Retinal mitochondrial DNA mismatch repair in the development of diabetic retinopathy, and its continued progression after termination of hyperglycemia. Invest. Ophthalmol. Vis. Sci., 2014; 55: 6960-7. [PubMed]
156: Estopinal CB , Chocron IM, Parks MB, Wade EA, Roberson RM, Burgess LG, Brantley. M Jr and Samuels DC. Mitochondrial haplogroups are associated with severity of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014; 55: 5589-95. [PubMed]
157: Kowluru RA, Kowluru V, Xiong Y, Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006; 41: 1191-6. [PubMed]
158: Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX. Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes. 2010; 59: 1528-38. [PubMed]
159: Satofuka S1, Ichihara A, Nagai N, Noda K, Ozawa Y, Fukamizu A, Tsubota K, Itoh H, Oike Y, Ishida S. (Pro)renin receptor-mediated signal transduction and tissue renin-angiotensin system contribute to diabetes-induced retinal inflammation. Diabetes.2009; 58: 1625-33. [PubMed]
160: Silva KC1, Rosales MA, Biswas SK, Lopes de Faria JB, Lopes de Faria JM. Diabetic retinal neurodegeneration is associated with mitochondrial oxidative stress and is improved by an angiotensin receptor blocker in a model combining hypertension and diabetes. Diabetes. 2009; 58: 1382-90. [PubMed]
161: Drel VR1, Xu W, Zhang J, Kador PF, Ali TK, Shin J, Julius U, Slusher B, El-Remessy AB, Obrosova IG. Poly(ADP-ribose)polymerase inhibition counteracts cataract formation and early retinal changes in streptozotocin-diabetic rats. Invest Ophthalmol Vis Sci. 2009; 50: 1778-90.4. [PubMed]
162: Fukumoto M1, Takai S, Ishizaki E, Sugiyama T, Oku H, Jin D, Sakaguchi M, Sakonjo H, Ikeda T, Miyazaki M. Involvement of angiotensin II-dependent vascular endothelial growth factor gene expression via NADPH oxidase in the retina in a type 2 diabetic rat model. Curr Eye Res. 2008; 33: 885-91. [PubMed]
163: Zheng L, Szabó C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004; 53: 2960-7. [PubMed]
164: Inoguchi T1, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000; 49: 1939-45. [PubMed]
165: Kern TS1, Tang J, Mizutani M, Kowluru RA, Nagaraj RH, Romeo G, Podesta F, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000; 41: 3972-8. [PubMed]
166: Nishikawa T1, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000; 404: 787-790. [PubMed]
167: Ganz MB1, Seftel A. Glucose-induced changes in protein kinase C and nitric oxide are prevented by vitamin E. Am J Physiol Endocrinol Metab. 2000; 278: E146-52. [PubMed]
168: Mohammad G, Siddiquei MM, Abu El-Asrar AM. Poly (ADP-ribose) polymerase mediates diabetes-induced retinal neuropathy. Mediators Inflamm. 2013; 2013: Article ID 510451. [PubMed]
169: Zhu Y, Zhang XL, Zhu BF, Ding YN. Effect of antioxidant N-acetylcysteine on diabetic retinopathy and expression of VEGF and ICAM-1 from retinal blood vessels of diabetic rats. Mol Biol Rep. 2012; 39: 3727-35. [PubMed]
170: Kowluru RA, Santos JM, Zhong Q. Sirt1, A Negative Regulator of Matrix Metalloproteinase-9 in Diabetic Retinopathy. Invest. Ophthalmol. Vis. Sci. 2014; 55: 5653-60. [PubMed]
171: Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, Spinowitz BS, Whittier FC, Wuerth JP; ACTION I Investigator Group. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004; 24: 32-40. [PubMed]
172: Keech AC1, Mitchell P, Summanen PA, O’Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d’Emden MC, Crimet DC,O’Connell RL, Colman PG; FIELD study investigators. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007; 370(9600):1687-97. [PubMed]
173: Gerss J, Hammes HP, Kampik A, Ulbig MW; RETIPON Study Group. Alpha-lipoic acid for the prevention of diabetic macular edema. Ophthalmologica. 2011; 226: 127-37. [PubMed]
174: PKC-DRS2 Group1, Aiello LP, Davis MD, Girach A, Kles KA, Milton RC, Sheetz MJ, Vignati L, Zhi XE. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology. 2006; 113: 2221-30. [PubMed]
175: Sheetz MJ, Aiello LP, Davis MD, Danis R, Bek T, Cunha-Vaz J, Shahri N, Berg PH. MBDL and MBCU Study Groups. The effect of the oral PKC β inhibitor ruboxistaurin on vision loss in two phase 3 studies. Invest Ophthalmol Vis Sci. 2013; 54: 1750-7. [PubMed]
176: Ellis EA1, Guberski DL, Somogyi-Mann M, Grant MB. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 2000; 28: 91-101. [PubMed]
177: Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005; 54: 1615-25. [PubMed]
177: Droge W. Free radicals and the physiological control of cell function. Physiol Rev. 2002; 83: 47–95. [PubMed]
178: Kern TS, Engerman RL. Capillary lesions develop in retina rather than cerebral cortex in diabetes and experimental galactosemia. Arch Ophthalmol. 1996; 114: 306-10. [PubMed]
179: Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008; 586: 4401-8. [PubMed]
180: Wolter, JR. Diabetic Retinopathy Amer J. Ophthalmol. 1961; 51: 1123-41. [PubMed]
181: Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997; 115: 1031-5. [PubMed]
182: Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998; 102: 783-91. [PubMed]
183: Takahashi H, Goto T, Shoji T, Tanito M, Park M, Chihara. Diabetes-associated retinal nerve fiber damage evaluated with scanning laser polarimetry. Amer J Ophthalmol. 2006; 142: 88-94. [PubMed]
184: Cho NC, Poulsen GL, Ver Hoeve JN, Nork TM. Selective loss of S-cones in diabetic retinopathy. Arch Ophthalmol. 2000; 118: 1393-400. [PubMed]
185: Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. 2004; 45: 3330-6. [PubMed]
186: Énzsöly A, Szabó A, Kántor O, Dávid C, Szalay P, Szabó K, Szél Á, Németh J, Lukáts Á. Pathologic alterations of the outer retina in streptozotocin-induced diabetes. Invest Ophthalmol Vis Sci. 2014; 55: 3686-99. [PubMed]
187: Ly A, Yee P, Vessey KA, Phipps JA, Jobling AI, and Fletcher EL. Early Inner Retinal Astrocyte Dysfunction during Diabetes and Development of Hypoxia, Retinal Stress, and Neuronal Functional Loss. Invest Ophthalmol Vis Sci. 2011; 52: 9316-9326. [PubMed]
188: Hombrebueno JR, Chen M, Penalva RG, Xu H1. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS One. 2014; 9: e97970. doi: 10.1371. [PubMed]
189: Drasdo N, Chiti Z, Owens DR, North RV. Effect of darkness on inner retinal hypoxia in diabetes. Lancet. 2002; 359(9325): 2251–2253. [PubMed]
190: Fortune B, Schneck ME, Adams AJ. Multifocal electroretinogram delays reveal local retinal dysfunction in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 1999; 40: 2638-51. [PubMed]
191: Ng JS, Bearse MA Jr, Schneck ME, et al. Local diabetic retinopathy prediction by multifocal ERG delays over 3 years. Invest Ophthalmol Vis Sci. 2008; 49: 1622-8. [PubMed]
192: Dean FM, Arden GB, Dornhorst A. Partial reversal of protan and tritan colour defects with inhaled oxygen in insulin dependent diabetic subjects. Br J Ophthalmol. 1997; 81: 27-30. [PubMed]
193: Kurtenbach A, Mayser HM, Jägle H, Fritsche A, Zrenner E. Hyperoxia, hyperglycemia, and photoreceptor sensitivity in normal and diabetic subjects. Vis Neurosci 2006; 23: 651-61. [PubMed]
194: Wong R, Khan J, Adewoyin T, Sivaprasad S, Arden GB, Chong V. The ChromaTest, a digital color contrast sensitivity analyzer, for diabetic maculopathy: a pilot study. BMC Ophthalmol. 2008; 8: 15. [PubMed]
195: Cringle SJ, Yu DY, Alder V, Su EN. Light and choroidal PO2 modulation of intraretinal oxygen levels in an avascular retina. Invest Ophthalmol Vis Sci. 1999; 40: 2307-13. [PubMed]
196: Harris A, Danis RP, Evans S, et al. Hyperoxia improves contrast sensitivity in early diabetic retinopathy. Br J Ophthalmol. 1996; 80: 209–13. [PubMed]
197: Van Dijk HW, Kok PH, Garvin M, et al. Selective loss of inner retinal layer thickness in type 1 diabetic patients with minimal diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009; 50: 3404-9. [PubMed]
198: Van Eeden PE, Tee LB, Lukehurst S, et al. Early vascular and neuronal changes in a VEGF transgenic mouse model of retinal neovascularization. Invest Ophthalmol Vis Sci. 2006; 47: 4638-45. [PubMed]
199: Lau JC and Linsenmeier RA. Increased intraretinal PO2 in short-term diabetic rats. Diabetes. 2014; 63.12: 4338-4342. [PubMed]
200: Puro DG. Diabetes-induced dysfunction of retinal Müller cells. Trans Am Ophthalmol Soc. 2002; 100: 339-52. [PubMed]
201: Ramsey DJ, Ripps H, Qian H. An electrophysiological study of retinal function in the diabetic female rat. Invest Ophthalmol Vis Sci. 2006; 47: 5116-24. [PubMed]
202: Castilho A, Ambrosio A, Hartveit E, and Veruki M. Disruption of a neural microcircuit in the rod pathway of the Mammalian Retina by Diabetes mellitus. J Neurosci. 2015; 35: 5422-5433. [PubMed]
203: Arden GB, Wolf JE, Collier J, Wolff C, Rosenberg M. Dark adaptation is impaired in diabetic before photopic loss can be seen. Can hypoxia contribute to diabetic retinopathy? In: Hollyfield JG (ed). Retinal Degenerative Diseases and Experimental Therapy. Plenum Press: New York, 1999, pp 305-325.
204: Arden GB, Sidman RL, Arap W and Schlingemann RO. Spare the rod and spoil the eye. Br J Ophthalmol. 2005; 89: 764-9. [PubMed]
205: Arden, GB, Wolf JE and Tsang Y. Does dark adaptation exacerbate diabetic retinopathy? Evidence and a linking hypothesis. Vision Res. 1998; 38: 1723-9. [PubMed]
206: Hagins WA, Penn RD, Yoshikami S. Dark current and photocurrent in retinal rods. Biophys J. 1970 10: 380-412. [PubMed]
207: Hodgkin AL, McNaughton PA, Nunn BJ, Yau KW. Effect of ions on retinal rods from Bufo marinus. J Physiol. 1984; 350: 649-680. [PubMed]
208: Neuroscience, 2nd edition Ed: Dale Purves, George J Augustine, David Fitzpatrick, et al. Sinauer Associates; 2001. In Chapter 11. Vision: The Eye: Phototransduction
209: Warburg O. Uber die Klassifizierung tierischer Gewebe nach ihrem Stoffwechsel. Biochem Z. 1927; 184: 484–488.
210: Warburg O, Posener K, Negelein E . Uber den Stoffwechsel der Carcinomzelle Biochemedische Zeitschrift 1924; 152: 309-43.
211: Linsenmeier RA. The effect of light and darkness on oxygen distribution and consumption in the cat retina. J Gen Physiol. 1986; 88: 521–542. [PubMed]
212: Daniele S, Daniele C. Aggravation of laser-treated diabetic cystoid macular edema after prolonged flight: a case report. Aviat Space Environ Med. 1995; 66: 440-2. [PubMed]
213: Khalifah RG, Bayners JW, Hudson BG, Amadorins. Novel post-Amadori inhibition of advanced glycation reactions. Biochem Biophys Res Commum. 1999; 257: 251-8. [PubMed]
214: Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006; 114: 597-605. [PubMed]
215: Chen M , Curtis TM, Stitt AW. Advanced glycation end products and diabetic retinopathy. Curr Med Chem. 2013; 20: 3234-40. [PubMed]
216: Du Y, Veenstra A, Palczewski K, Kern TS. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. PNAS. 2013; 110: 16586-16591. [PubMed]
217: Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983; 219: 983-985 [PubMed]
218: Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res. 2013; 34:19-48. [PubMed]
219: Mohammad G, Siddiquei MM, Othman A, Al-Shabrawey M, Abu El-Asrar AM. High-mobility group box-1 protein activates inflammatory signaling pathway components and disrupts retinal vascular-barrier in the diabetic retina. Exp Eye Res. 2013; 107: 101-9. [PubMed]
220: Milne R, Brownstein S. Advanced glycation end products and diabetic retinopathy. Amino Acids. 2013; 44: 1397-407. [PubMed]
221: Zong H, Ward M, Stitt AW. AGEs, RAGE, and diabetic retinopathy. Curr Diab Rep. 2011; 11: 244-52. [PubMed]
222: Stitt AW. AGEs and diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010; 51: 4867-74. [PubMed]
223: Madsen-Bouterse SA, Kowluru RA. Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Rev Endocr Metab Disord 2008; 9: 315–327. [PubMed]
224: Pournaras CJ, Riva CE, Tsacopoulos M, Strommer K. Diffusion of O2 in the retina of anesthetized miniature pigs in normoxia and hyperoxia. Exp Eye Res. 1989; 49: 347-60. [PubMed]
225: Frank RN. Visual fields and electroretinography following extensive photocoagulation. Arch Ophthalmol. 1975; 93: 591-8. [PubMed]
226: Grunwald JE, Riva CE, Brucker AJ, Sinclair SH, Petrig BL. Altered retinal vascular response to 100% oxygen breathing in diabetes mellitus. Ophthalmology. 1984; 91: 1447-52. [PubMed]
227: Feke GT, Buzney SM, Ogasawara H, Fujio N, Goger DG, Spack NP, Gabbay KH. Retinal circulatory abnormalities in type 1 diabetes. Invest Ophthalmol Vis Sci. 1994; 35: 2968-75. [PubMed]
228: Bursell SE, Clermont AC, Kinsley BT, Simonson DC, Aiello LM, Wolpert HA. Retinal blood flow changes in patients with insulin-dependent diabetes mellitus and no diabetic retinopathy. Invest Ophthalmol Vis Sci. 1996; 37: 886-97. [PubMed]
229: Ahmed J, Hatchell DL et al. Retinal hypoxia in long-term diabetic cats. Invest Ophthalmol Vis Sci 1998; 39: 1647-1657.
230: Linsenmeier RA, Braun RD, McRipley MA, Padnick LB, Ahmed J, Hatchell DL, McLeod DS, Lutty GA. Retinal hypoxia in long-term diabetic cats. Invest Ophthalmol Vis Sci. 1998 ;39: 1647-57. [PubMed]
231: Minot, CS. The Problem of Age, Growth and Death. The Popular Science Monthly.
232: Yu DY, Cringle SJ. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Ret. Eye Res. 2001; 20: 175-208. [PubMed]
233: Yu DY, Cringle SJ, Alder VA, Su EN. Intraretinal oxygen distribution in rats as a function of systemic blood pressure. Am J Physiol. 1994; 267: H2498-507. [PubMed]
234: Yu DY Cringle SJ. Outer retinal anoxia during dark adaptation is not a general property of mammalian retinas. Comp Biochem Physiol 2002; A132: 47-52. [PubMed]
235: Yu DY, Cringle SJ. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res. 2001; 20: 175-208. [PubMed]
236: Birol G, Wang S, Budzynski E, Wangsa-Wirawan ND, Linsenmeier RA. Oxygen distribution and consumption in the macaque retina. Am J Physiol Heart Circ Physiol. 2007; 293: H1696-704. [PubMed]
237: Tofts PS, Porchia A, Jin Y, Roberts R, Berkowitz BA. Toward clinical application of manganese-enhanced MRI of retinal function. Brain Res Bull. 2010; 81: 333-8. [PubMed]
238: de Gooyer TE, Stevenson KA, Humphries P, Simpson DA, Curtis TM, Gardiner TA, Stitt AW. Rod photoreceptor loss in Rho-/- mice reduces retinal hypoxia and hypoxia-regulated gene expression Invest Ophthalmol Vis Sci. 2006; 47: 5553-60. [PubMed]
239: de Gooyer TE1, Stevenson KA, Humphries P, Simpson DA, Gardiner TA, Stitt AW. Retinopathy is reduced during experimental diabetes in a mouse model of outer retinal degeneration. Invest Ophthalmol Vis Sci. 2006; 47: 5561-8. [PubMed]
240: Arden GB. The absence of diabetic retinopathy in patients with retinitis pigmentosa: implications for pathophysiology and possible treatment. Brit J Ophthalmol 2001; 85: 366-370. [PubMed]
241: Smith PR, Bain SC, Good PA, et al. Pigmentary retinal dystrophy and the syndrome of maternally inherited diabetes and deafness caused by the mitochondrial DNA 3243 tRNA(Leu) A to G mutation. Ophthalmology. 1999; 106: 1101-1108. [PubMed]
242: Holmes-Walker DJ, Mitchell P, Boyages SC. Does mitochondrial genome mutation in subjects with maternally inherited diabetes and deafness decrease severity of diabetic retinopathy? Diabet Med. 1998; 15: 946-952. [PubMed]
243: Marsh S, Nakhoul FM, Skorecki K, Rubin A, Miller BP, Leibu R, Levy NS, Levy AP. Hypoxic induction of vascular endothelial growth factor is markedly decreased in diabetic individuals who do not develop retinopathy. Diabetes Care. 2000; 23: 1375–1380. [PubMed]
244: Harris A, Arend O, Danis RP, Evans S, Wolf S, Martin BJ. Hyperoxia improves contrast sensitivity in early diabetic retinopathy. Br J Ophthalmol 1996; 80: 209-213. [PubMed]
245: Nguyen QD, Shah SM, Van Anden E, Sung JU, Vitale S, Campochiaro PA. Supplemental oxygen improves diabetic macular oedema: a pilot study. Invest Ophthalmol Vis Sci. 2004; 45: 617-624. [PubMed]
246: Shiba T, Sato Y, Takahashi M. Relationship between proliferative diabetic retinopathy and sleep-disordered breathing. Nippon Ganka Gakkai Zasshi. 2007; 111: 899-904. [PubMed]
247: Shiba T, Sato Y, Takahashi M. Relationship between diabetic retinopathy and sleep-disordered breathing. Am J Ophthalmol. 2011; 147: 1017-1021. [PubMed]
248: West SD, Groves DC, Lipinski HJ, et al. The prevalence of retinopathy in men with Type 2 diabetes and obstructive sleep apnoea. Diabet Med. 2010; 27: 423-430. [PubMed]
249: Kosseifi S, Bailey B, Price R, Roy TM, Byrd RP Jr, Peiris AN. The association between obstructive sleep apnea syndrome and microvascular complications in well-controlled diabetic patients. Mil Med. 2010; 175: 913-916. [PubMed]
250: Mason RH, West SD, Kiire CA, et al. High prevalence of sleep disordered breathing in patients with diabetic macular edema. Retina. 2012; 32: 1791-8. [PubMed]
251: Arden GB, Sivaprasad S. Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr Diabetes Rev. 2011; 7: 291-304. [PubMed]
252: Arden GB, Sivaprasad S. The pathogenesis of early retinal changes of diabetic retinopathy. Doc Ophthalmol. 2012; 124: 15-26. [PubMed]
253: Arden GB, Gǔndǔz MK, Kurtenbach A, Volker M, Zrenner E, Gǔndǔz SB et al. Preliminary trial to determine whether prevention of dark adaptation affects the course of early diabetic retinopathy. Eye 2010; 24: 1149-1155. [PubMed]
254: Arden GB, Jyothi S, Hogg CH, Lee YL, and Sivaprasad S. Regression of early diabetic macular oedema is associated with prevention of dark adaptation. Eye 2011; 25: 1546-1554. [PubMed]
255: Sivaprasad S, Arden G, Prevost AT, Crosby-Nwaobi R, Holmes H, Kelly J, Murphy C, Rubin G, Vasconcelos J, Hykin P. A multicentre phase III randomised controlled single-masked clinical trial evaluating the clinical efficacy and safety of light-masks at preventing dark-adaptation in the treatment of early diabetic macular oedema (CLEOPATRA): study protocol for a randomised controlled trial. Trials. 2014; Nov 22;15: 458. [PubMed]
256: McKeague C Margrain TH Baliey C Binns AM Low-level night-time therapy for age-related macular degeneration (Alight): study protocol for a randomised controlled trial. Trials 2014; 15: 246 [PubMed]
257: Chakravaty U, Harding SP, Rogers et al. Ranibizumab versus bevacizumab to treat neovascular AMD one-year findings from the IVAN randomized trial. Ophthalmology 2012; 119: 1399-411. [PubMed]
258: Rosenfeld PJ, Rich RM, Lalwani GA. Ranibizumab: Phase III clinical trial results. Ophthalmol Clin North Am. 2006; 19: 361-72. [PubMed]
259: Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol. 2011; 8: 228-36. [PubMed]
260: Bressler NM, Varma R, Doan QV, Gleeson M, Danese M, Bower JK, Selvin E, Dolan C, Fine J, Colman S, Turpcu A. Underuse of the health care system by persons with diabetes mellitus and diabetic macular edema in the United States. JAMA Ophthalmol. 2014; 132: 168-73. [PubMed]
261: Rosenfeld PJ, Rich RM, Lalwani GA. Ranibizumab: Phase III clinical trial results. Ophthalmol Clin North Am. 2006; 19: 361-72. [PubMed]
262: Bloch SB, Larsen M. Treatment of wet age-related macular degeneration with vascular endothelial growth factor. A survey of a Cochrane review Ugeskr Laeger. 2009 16; 171: 3435-7. [PubMed]
263: Mitchell P. Cytokines in neovascular age-related macular degeneration: fundamentals of targeted combination therapy. Br J Ophthalmol. 2011; 95: 1631-7. [PubMed]
264: Stewart MW. The expanding role of vascular endothelial growth factor inhibitors in ophthalmology. Mayo Clin Proc. 2012; 87): 77-88. [PubMed]
265: Wolf A, Kampik A. Efficacy of treatment with ranibizumab in patients with wet age-related macular degeneration in routine clinical care: data from the COMPASS health services research. Graefes Arch Clin Exp Ophthalmol. 2014; 252: 647-55. [PubMed]
266: Micieli JA, Surkont M, Smith AF. A systematic Analysis of the Off-Label Use of Bevacizumab for Severe Retinopathy of Prematurity. Am J Ophthalmol. 2009; 148: 536-43. [PubMed]
267: Assari T Chronic Granulomatous Disease; fundamental stages in our understanding of CGD Medical Immunology e2006; 5: 4-31. [PubMed]
268: Kita K, Konishi K, Anraku Y. Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol Chem. 1984; 259: 3375-3381. [PubMed]
269: www.nice.org.uk/guidance/ta274/resources
270. Ramsey DJ, Ripps H, Qian H. Streptozotocin-induced diabetes modulates GABA receptor activity of rat retinal neurons. Exp Eye Res. 2007; 85 :413-22. Epub 2007 Jun 19. [PubMed]