What is glaucoma?
David Križaj
Abstract
Glaucoma, a leading cause of blindness in the world, can be challenging to diagnose because symptoms often appear at late stage of the disease, and challenging to treat because of the irreversible loss of retinal neurons. The term encompasses a heterogenous group of diseases that are characterized by altered biomechanics of anterior and posterior eye. These diseases tend to manifest as stiffening of the trabecular meshwork and increased production/reduced drainage of aqueous humor, together with retinal inflammation associated with activated microglia, Müller cells and astrocytes, and degeneration of retinal ganglion cells. In general, the phenotype is caused by a conjunction of risk factors such as age, family history, ethnic origin, high myopia, vascular disease and intraocular pressure (IOP). Current treatments are limited to lowering and stabilizing IOP, indicating that glaucoma is principally a disease of ocular mechanotransduction. Although the events that lead from IOP elevations to ganglion cell damage are not well defined, a body of recent work has pointed at mechanotransducing TRPV4, piezo and TREK-1 channels and immune mechanisms as key pressure targets in trabecular meshwork, ciliary body, retinal ganglion, endothelial cells and glia. Data indicate that dysfunctions of mechanotransduction mechanisms within the ganglion cell soma-dendrite, Müller cells and microglia may precede axonal degeneration, cupping of the optic nerve head and visual field damage which over the years have represented the golden standard of glaucoma diagnosis. Collectively, these recent studies predict the development of new therapies and diagnostic strategies to be contingent upon systematic delineation of molecular mechanisms that sense and transduce pressure in the eye. Thus, neuroprotection, a currently elusive goal, may be achieved via parallel suppression of pressure sensing in the anterior eye, retinal neurons, blood vessels and glia in order to achieve normalization of neuron-glia-blood vessel interactions, and the attendant reduction in immune activation.
Introduction
Glaucoma, a group of optic neuropathies characterized by progressive degeneration of retinal ganglion cells (RGCs), represents the leading cause of irreversible blindness in the developed world. Typical symptoms include gradual loss of peripheral (side) vision that is followed by progressive loss of central vision. If left untreated, glaucoma can progress to complete blindness. Because the damage to the eye is slow and painless, only half of the carriers are aware of the disease and irreparable harm often takes place long before diagnosis. Glaucoma is generally identified by abnormal regulation of intraocular pressure (IOP) and/or pathological mechanosensitivity of ocular cells but the relationship between mechanical forces and the disease remains enigmatic and a matter of considerable academic, clinical and economic interest. The proportion of blindness attributable to glaucoma varies considerably from the lowest values in South Asia to high prevalence in sub-Saharan Africa (1). By 2020, the disease is expected to affect >76 million people, with an estimated 3 million afflicted in the United States alone. There is currently no cure because retinal neurons that die do not regenerate, however, progression of the disease may be slowed with drugs that lower intraocular pressure (IOP). The economic burden (medical costs, assistance programs in the U.S. estimated at ~$3 billion/year) doubles when considering indirect costs associated with productivity loss, physical consequences (increase in hip fractures; increase in family care) and decreased quality of life. Given that early detection of glaucoma, initiation and adherence to treatment strongly correlate with socioeconomic status (2), there is much room for improvement in providing care.
First descriptions of glaucoma-like eye disease conditions are from ancient Greece. Aristotle and Hippocrates (~500 BC) used the term glaukos (γλαυκóς) to describe a late-stage condition characterized by the gray-green-blue hue of the pupil (3). In a remarkable approximation of close angle glaucoma, Galen (c.129-199 AD) linked this glaucous hue to deficient levels of aqueous humor, a shallow anterior chamber and/or anteriorly prominent lens. The “Book of Hippocratic Treatment” by Al-Tabari, a 10th century Arab physician, may have provided some of the first references to intraocular pressure yet medieval Europe continued to categorize glaucoma as an incurable green (viriditas) cataract (“waterfall”) and “excessive hardness” of the lens (4). Confusion ensued when removal of the opaque lens turned out to improve vision but the link to IOP was resurrected in the mid- 19th century by Franciscus Cornelius Donders (Dutch) who developed the first tonometer. Albrecht von Graefe (German) and Antoine-Pierre Demours (French) linked the disease to the iridocorneal dysfunction however glaucoma was defined and canonized by William Mackenzie (Scottish) in his “Practical Treatise of the Diseases of the Eye” (1853) (5). The medieval designation of glaucoma as “green cataract” continues to persist in some languages (such as Slovenian).
A distinctive feature of glaucoma is the obligatory association between biological processes that take place in two distinct locations within the eyeball (Figure 1). A pressure gradient in the anterior eye contributes – through mechanisms that are still unclear – to optic neuropathy in the posterior eye (retina). To adapt to increased mechanical forces, trabecular meshwork cells in the anterior eye, retinal ganglion cells and glia undergo structural and functional changes known as glaucomatous remodeling. This process involves includes many mechanisms at nanoscale (gene expression), mesoscale (changes in enzymes, ion channels, cytoskeletal proteins, receptors and secreted molecules) to macroscale (neuropathy) levels with the final common consequence of the lost retinal output to the brain (Figure 1).
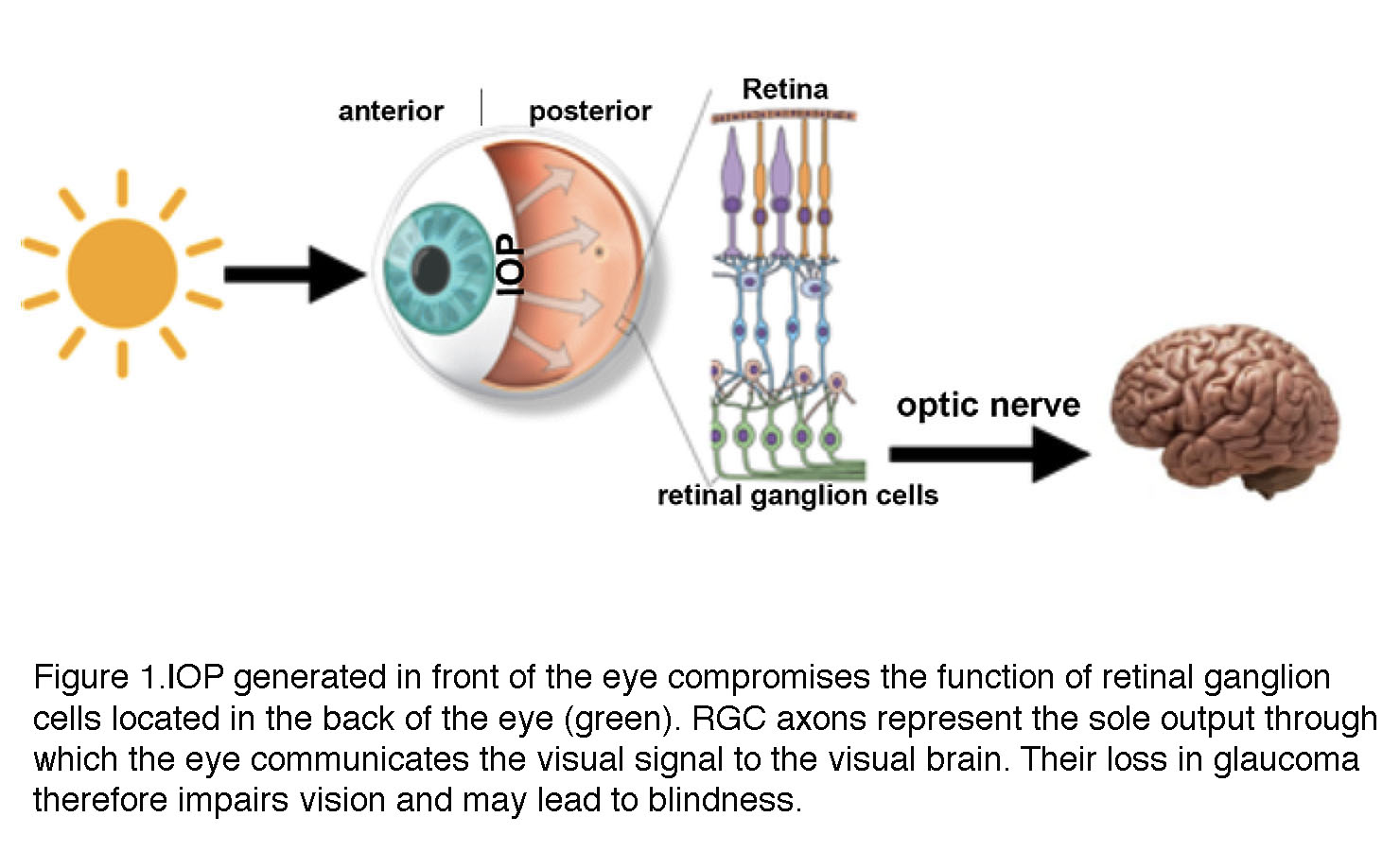
Figure 1. IOP generated in front of the eye (anterior) compromises the function of retinal ganglion cells (green, RGC) located in the back of the eye (posterior). RGC axons traveling through the optic nerve represent the sole output through with the eye communicates the visual signal to the visual areas of the brain. Axonal loss in glaucoma therefore impairs vision and may lead to blindness.
Among the broad range of risk factors that influence the likelihood of developing glaucoma are age, ethnic origin, family history, corneal thickness, low blood pressure, cerebrospinal fluid pressure, intraocular pressure and vascular dysregulation. The multiplicity of these stressors, some of which are IOP-independent, underlies the differences in the rate of glaucomatous progression and response to treatment across individual patients.
It is important to note that criteria for diagnosing glaucoma cannot yet identify molecular and physiological markers of the disease and that the lack of a structure-function framework that unifies the bewildering constellation of glaucoma risk factors and phenotypes represents a significant unmet need in both the diagnosis and treatment of glaucoma. The widely used textbook definition of glaucoma as “an optic neuropathy defined by ‘cupping’ of the optic nerve head” is an antiquated description of a late stage condition at which early warnings stages of IOP- induced changes in retinal neuronal excitability, synaptic remodeling, neuroinflammation and altered light responsiveness have already taken place. It also fails to bring insight into the ongoing debate whether glaucoma is one disease with a continuum across phenotypes or a group of diseases with distinct genetic origin and signaling mechanisms. This review presents a bird’s eye overview of some of the current topics of controversy and/or areas where knowledge is currently lacking. It reflects the author’s personal views and also regrets that the large majority of the relevant studies could not be cited directly (although many are referenced in the secondary literature).
Diagnosis of glaucoma.
The current clinical diagnosis of glaucoma is based on the measurements of IOP, visual function and retinal structure. Stereoscopic “slit lamp” ophthalmoscopy and optical coherence tomography (OCT) check for the “cupping” (concavity) of the optic nerve head and degeneration of the retinal nerve fiber layer (composed of RGC axons); perimetry documents the functional damage in vision by testing for arcuate scotoma (regional loss of peripheral vision), and tonometry is used to measure IOP (6, 7). Patients at risk for the primary angle-closure type of disease (see below) are additionally examined with gonioscopy, which bounces light into the eye using a specialized lens. An example of the ophthalmoscopic examination of the interior surface (“fundus”) of the eye is shown in Figure 2 and Figure 3. An “excavated” or “cupped” optic nerve head in Figure 2 is associated with reduced vascularization of the foveal region whereas Figure 3 shows a typical late-stage example, with magnified views of the optic nerve head of a normal patients retina (left) and the same view of a patient’s retina with severe glaucoma (right).
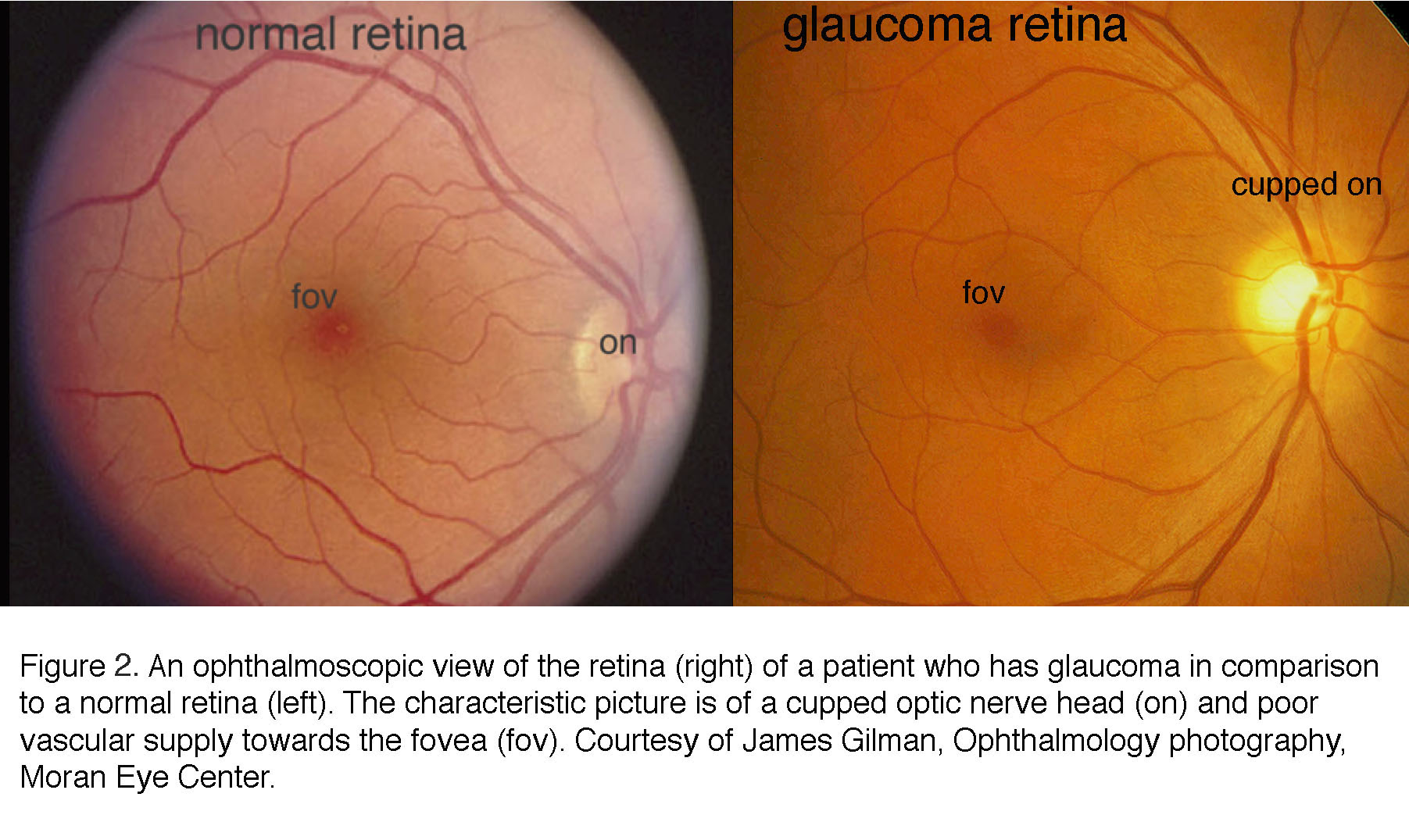 Figure 2. An ophthalmoscopic view of the retina (right) of a patient who has glaucoma, in comparison to a normal retina (left). The characteristic picture is of a cupped optic nerve head (on) and poor vascular supply towards the fovea (fov). Courtesy of James Gilman, Ophthalmology photography, Moran Eye Center.
Figure 2. An ophthalmoscopic view of the retina (right) of a patient who has glaucoma, in comparison to a normal retina (left). The characteristic picture is of a cupped optic nerve head (on) and poor vascular supply towards the fovea (fov). Courtesy of James Gilman, Ophthalmology photography, Moran Eye Center.
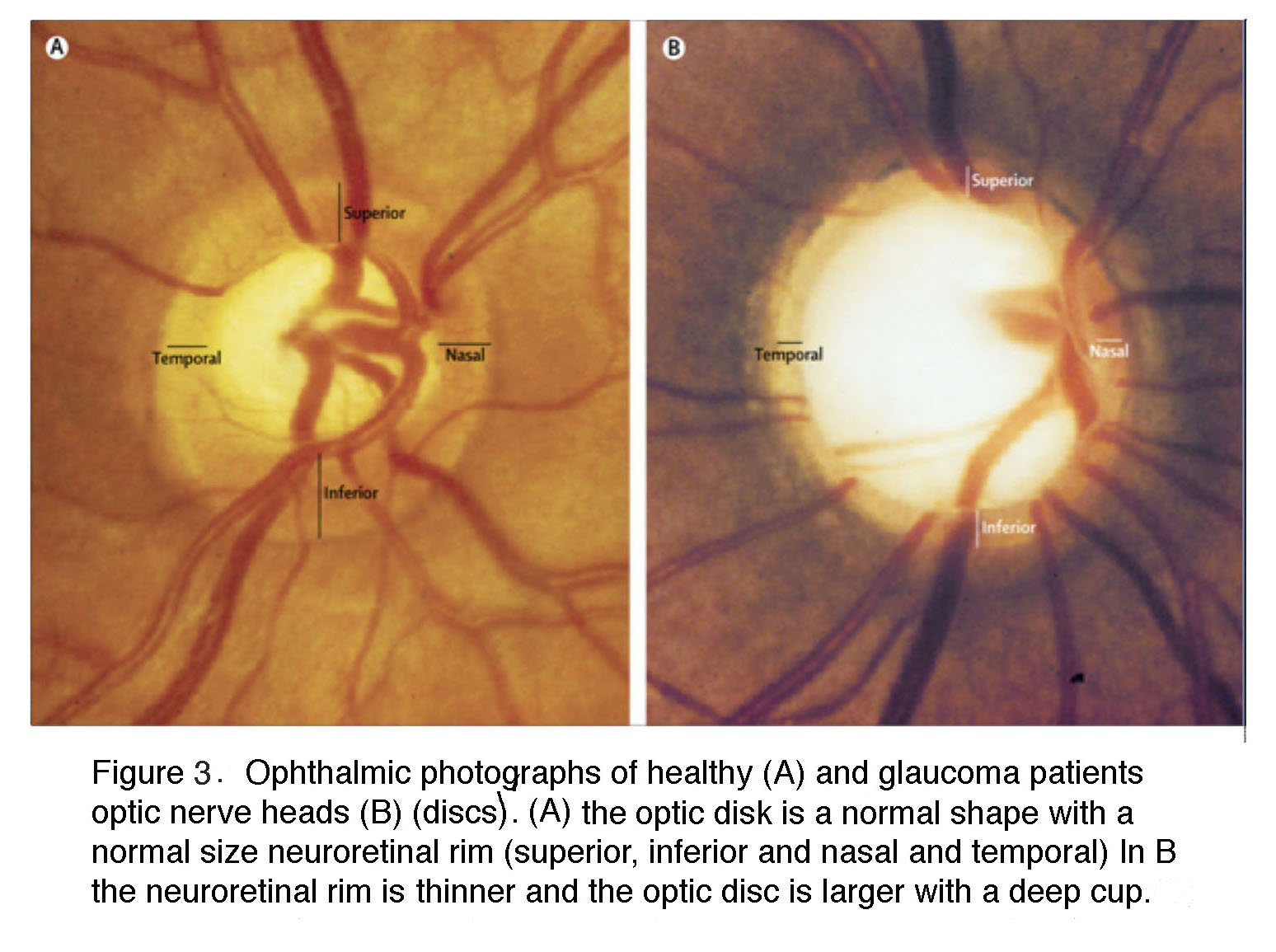
Figure 3. Ophthalmic photographs of healthy (A) and a glaucoma patient optic disks (B). The optic disk is the optic nerve head. (A) The optic disk is a normal shape, with a normally sized neuroretinal rim (superior, inferior, nasal and temporal). In (B) the neuroretinal rim is thinner, and the optic disk is larger, with a deep cup.
In the glaucomatous eye the optic nerve head appears larger because the neuroretinal rim is narrower and the center is cupped. In fundus photos of advanced glaucoma, degeneration of ganglion cell axons can be seen by the appearance of remaining axons in so called “wedge-shapes” that differ from the picture of healthy ganglion cell axons radiating in superior and inferior arches to the optic nerve head (Figure 4).
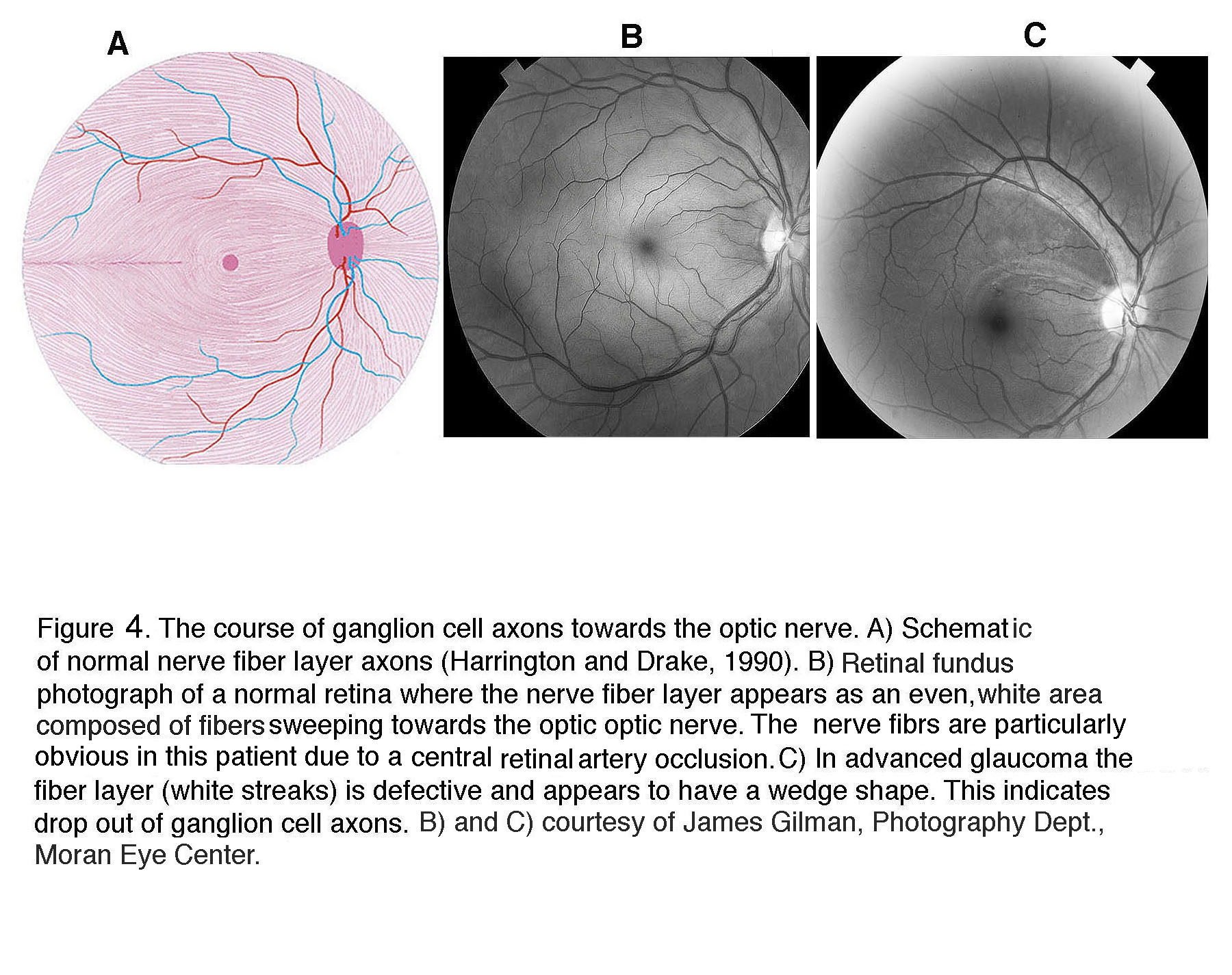 Figure 4. The course of ganglion cell axons towards the optic nerve head. (A) Schematic of normal nerve fiber layer axons (Harrington and Drake, 1990). (B) Retinal fundus photograph of a normal retina where the nerve fiber layer appears as an even, white area composed of fibers sweeping towards the optic nerve head. The nerve fibers are particularly obvious in this patient due to a central retinal artery occlusion. (C) In advanced glaucoma the fiber layer (white streaks) is defective and appears to have a wedge shape. This indicates drop out of ganglion cell axons. (B) and (C) courtesy of James Gilman, Photography Department, Moran Eye Center.
Figure 4. The course of ganglion cell axons towards the optic nerve head. (A) Schematic of normal nerve fiber layer axons (Harrington and Drake, 1990). (B) Retinal fundus photograph of a normal retina where the nerve fiber layer appears as an even, white area composed of fibers sweeping towards the optic nerve head. The nerve fibers are particularly obvious in this patient due to a central retinal artery occlusion. (C) In advanced glaucoma the fiber layer (white streaks) is defective and appears to have a wedge shape. This indicates drop out of ganglion cell axons. (B) and (C) courtesy of James Gilman, Photography Department, Moran Eye Center.
In general, functional loss can be detected once >30% of RGCs have died, although patients and animals may not notice that their vision is compromised until much later. Figure 5 shows that RGCs are the main neuronal cell type lost in a non-human primate retina with induced glaucoma. Macaque retinas with induced glaucoma (8) show substantial loss of RGCs following chronic IOP elevation (Figure 5A) but show visual sensitivity that is compromised only after the monkeys lost ~50% of their RGCs (Figure 5B).
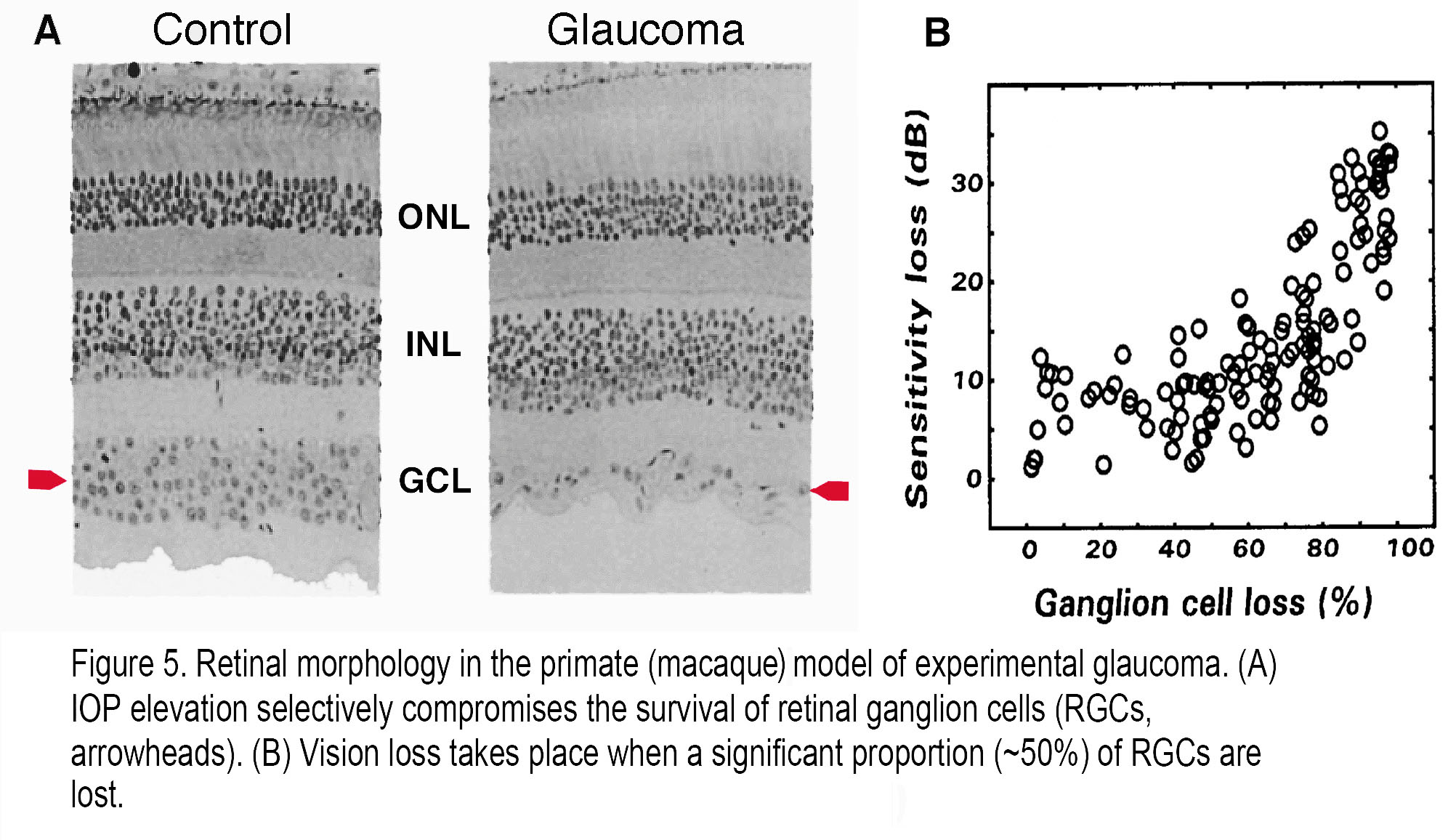 Figure 5. Retinal morphology in the primate (macaque) model of experimental glaucoma. (A) IOP elevation selectively compromises the survival of retinal ganglion cells (RGCs, arrowheads). (B) Vision loss takes place when a significant proportion (~50%) of RGCs are lost.
Figure 5. Retinal morphology in the primate (macaque) model of experimental glaucoma. (A) IOP elevation selectively compromises the survival of retinal ganglion cells (RGCs, arrowheads). (B) Vision loss takes place when a significant proportion (~50%) of RGCs are lost.
Patients with glaucoma often describe experiencing colored “haloes” that may reflect corneal epithelial edema and transient compression of the ONH. Many anecdotal cases link strong emotional trauma to acute manifestation of the disease however the relationship between the emotional/anxiety states and glaucoma remains to be systematically evaluated. In contrast to the wealth of studies in non-human primates, there have been relatively few histological investigations of glaucomatous human retinas. One example is shown in a 2003 study by Pavlidis and colleagues (Figure 6) (9). Like the macaque (Figure 5), few ganglion cells remained in the human retina with advanced glaucoma. The rare ganglion cells that were stained are atrophied, with few dendritic branches and loss of bushiness (Figure 6, panel K), shrunken cell bodies and notably beaded axons running in the nerve fiber layer (Figure 6, panels B,C,E, F and H, I) compared with normal midget ganglion cells (Figure 6. panels A, D, G and J). Larger cell types such as parasol ganglion cells were similarly affected in the advanced glaucomatous retina (9). For ganglion cell types in human retina see “Primate ganglion cells” in Morphology and circuitry of ganglion cells section in Webvision. For rodent models, see later in this chapter and review by Nickells et al. (2012) (10).
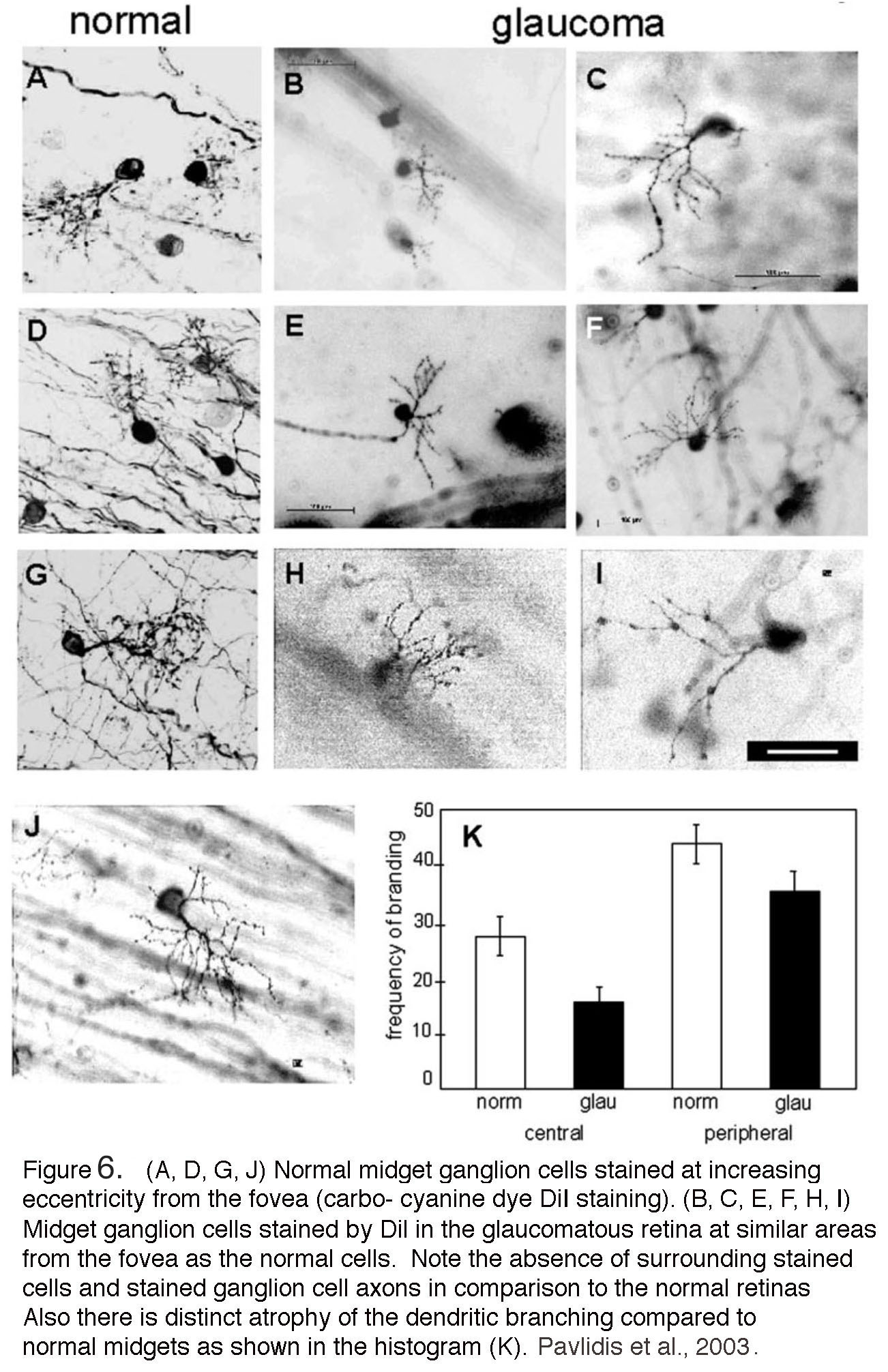 Figure 6. (A, D, G, J) Normal midget ganglion cells stained at increasing eccentricity from the fovea. Carbo-cyanine dye (DiI) staining. (B, C, E, F, H, I) Midget ganglion cells stained by DiI in the glaucomatous retina at similar distances from the fovea as the normal cells. Note the absence of surrounding stained cells and stained ganglion cell axons in comparison to the normal retinas. Also, there is distinct atrophy of the dendritic branching compared to normal midgets as shown in the histogram (K) (From Pavilidis et al, 2003).
Figure 6. (A, D, G, J) Normal midget ganglion cells stained at increasing eccentricity from the fovea. Carbo-cyanine dye (DiI) staining. (B, C, E, F, H, I) Midget ganglion cells stained by DiI in the glaucomatous retina at similar distances from the fovea as the normal cells. Note the absence of surrounding stained cells and stained ganglion cell axons in comparison to the normal retinas. Also, there is distinct atrophy of the dendritic branching compared to normal midgets as shown in the histogram (K) (From Pavilidis et al, 2003).
Types of glaucoma
“Primary” glaucomas show optic neuropathy in the presence or absence of elevated IOP (i.e., the cause of the disease is not known) whereas “secondary” glaucomas are defined by known pathological mechanisms that elevate IOP above normal. Both types of disease have in common dysfunction of the anterior eye, symptoms (RGC loss and optic neuropathy) and treatment (IOP lowering). Diagnosis continues to be a problem, as all too often glaucoma is identified after many RGCs (green cells in Figure 1) had been irretrievably lost. The most common forms of glaucoma do not show Mendelian inheritance, yet genetic and racial background is clearly important as ethnic groups show striking differences in the prevalence of types of glaucoma. For example, primary open-angle glaucoma (POAG) is widespread in Africa (4.2%) whereas primary angle closure glaucoma (PACG) is more common in Asia (1.1%). With respect to the population sizes, however, Asians still account for the majority (~53%) of worldwide POAG cases (2, 11). In general, gene mutations with large biological effects (i.e., optic neuropathy) are rare whereas gene variants associated with smaller effects are common (12) (see below). Treatment for all forms of glaucoma is limited to pharmacological lowering of IOP (the sole treatable risk factor) and, in rare cases, anterior eye surgery and laser photo/thermocoagulation but there are no treatments for neuronal degeneration (reviewed in (13-15)).
Primary open-angle glaucoma (POAG)
POAG is the most common form of glaucoma that accounts for >80% of cases in the Western hemisphere. The ‘angle’ refers to the iridocorneal cleft between the iris and the cornea which is “open” in normal eye and POAG (Figure 7A & Figure 7B) and offers access to aqueous humor flowing into the trabecular meshwork and Schlemm’s canal. Clinically, POAG is diagnosed by the open angle, IOP > 21 mm Hg and loss of visual field associated with the degeneration of ganglion cell axons (“optic neuropathy”) (16). While relatively uncommon under the age 50, POAG strikes nearly 8% of Americans over 80; it is about five times more common among African Americans than Caucasians, with earlier onset and faster time-course of progression in individuals with African heritage (17). On the other hand, people with European ancestry are more vulnerable compared to Africans or Asians when assessing the odds ratio of POAG per decade (18). The prevalence seems to be higher in men. In most cases of POAG, the disease may progress to total blindness without the patients experiencing any pain or discomfort, which however may occur when the rate of tension is very high, and IOP exceeds ~50 mm Hg.
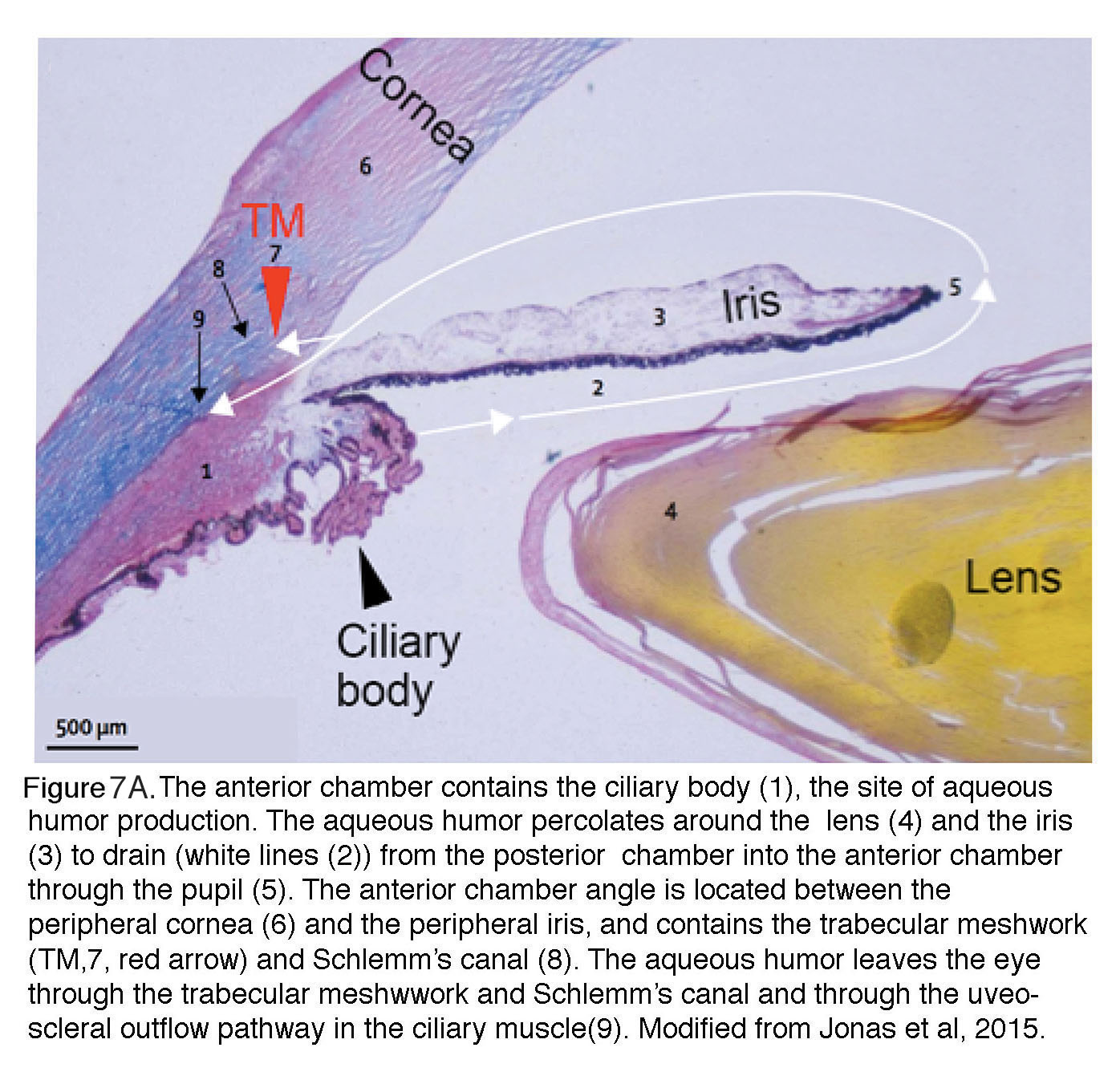 Figure 7A. The anterior chamber contains the ciliary body 1, the site of aqueous humor production. The aqueous humor percolates around the lens 4 and the iris 3 to drain (white lines 2) from the posterior chamber into the anterior chamber through the pupil 5. The anterior chamber angle is located between the peripheral cornea 6 and the peripheral iris, and it contains the trabecular meshwork (TM 7 red arrow) and Schlemm’s canal 8. The aqueous humor leaves the eye through the trabecular meshwork and Schlemm’s canal, and through the uveo-scleral outflow pathway in the ciliary muscle 9. Modified from Jonas et al, 2017 (13).
Figure 7A. The anterior chamber contains the ciliary body 1, the site of aqueous humor production. The aqueous humor percolates around the lens 4 and the iris 3 to drain (white lines 2) from the posterior chamber into the anterior chamber through the pupil 5. The anterior chamber angle is located between the peripheral cornea 6 and the peripheral iris, and it contains the trabecular meshwork (TM 7 red arrow) and Schlemm’s canal 8. The aqueous humor leaves the eye through the trabecular meshwork and Schlemm’s canal, and through the uveo-scleral outflow pathway in the ciliary muscle 9. Modified from Jonas et al, 2017 (13).
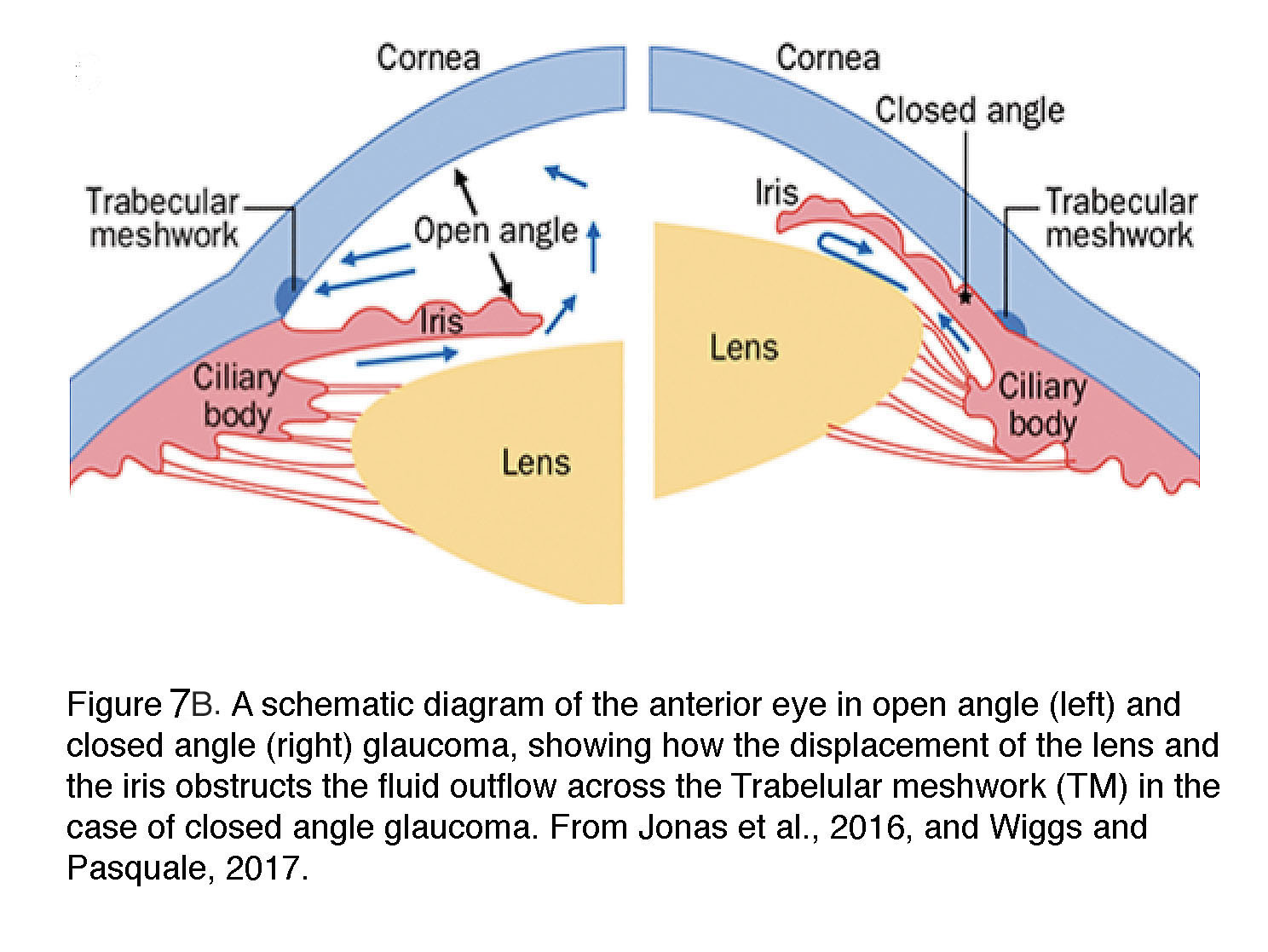 Figure 7B. A schematic diagram of the anterior eye in open angle (left) and closed angel (right) glaucoma, showing how the displacement of the lens and the iris obstructs the fluid outflow across the Trabecular meshwork (TM) in the case of closed angle glaucoma. From Wiggs and Pasquale, 2017 (12).
Figure 7B. A schematic diagram of the anterior eye in open angle (left) and closed angel (right) glaucoma, showing how the displacement of the lens and the iris obstructs the fluid outflow across the Trabecular meshwork (TM) in the case of closed angle glaucoma. From Wiggs and Pasquale, 2017 (12).
A key, heritable OAG risk factor is IOP, which varies between ~7 and >20 mm Hg. Every 1 mm Hg increase in baseline elevates the risk of developing the disease by 10-14% over any 5 – 9 -year period (19). Untreated eyes with IOP in the range of 21 – 25 mm Hg show ~14.4 years to progress from early to late stage of POAG but the lag period is shortened to ~2.9 years at IOP >30 mm Hg. Individuals with myopia are at risk for both elevated IOP and POAG, possibly due to weaker connective tissue support (18). It is important to note that the disease is unpredictable as there is a substantial fraction of patients with elevated IOP who show no signs of axonal degeneration, visual field defects and vision loss.
POAG has a clear genetic foundation, with heritability amongst twins ~0.95, and siblings of affected individuals having nearly an eight-fold increase risk of developing the disease. In contrast to macular degeneration (AMD), in which genome-wide studies (GWAS) identified ~60% of responsible genes, known POAG sequences comprise less than 10% of known cases. These include ~50 gene loci, with mutations in three Mendelian genes – myocilin (MYOC), optineurin (OPTN) and TANK binding kinase (TBK1) accounting for ~5% of POAG cases with little influence from other genes or environmental factors (12, 20). MYOC is a secreted calcium-binding protein with no known function, and is expressed in TM cells, retinal pigment epithelium and astrocytes. Mice and humans that lack or overexpress MYOC have normal aqueous drainage and do not develop high IOP whereas mice expressing the human Tyr437His mutation in the olfactomedin domain develop elevated IOP and optic nerve damage that mirrors human glaucoma (21). Carriers of MYOC mutations develop glaucoma in ~90% of cases, presumably due to deranged protein-protein interactions, intracellular accumulation of misfolded proteins and ER/endosomal stress (12). OPTN encodes a scaffold protein that has at least 23 binding partners, 8 protein binding sites and likely roles in multiple signaling pathways, including autophagy and vesicle trafficking in TM cells and RGCs (22).
A widely shared view of POAG is that of a multifactorial disease in which multiple genes, protein and lipid signaling pathways are dysregulated, but may also be influenced by environmental stressors and epigenetic mechanisms. The molecular “interactomes” that underpin pressure-sensitive and insensitive aspects of POAG remain to be fully characterized with potential clues from recent genome-wide association study (GWAS) analyses of adult-onset POAG loci including genes that encode cholesterol-binding caveolin proteins CAV1/2, ABCA1 (ATP-binding cassette transporter 1), TGFBR3 (transforming growth factor beta receptor 3), FOXC1 (forkhead box C1), ELOVL5 (elongation of long chain fatty acids family member 5), ATOH7 (atonal homolog 7), CYP1B1 (a cytochrome P450 enzyme) and ATXN2 (ataxin isoform 2) proteins. On their own, these proteins may be harmless – and/or independent of IOP – but may accumulate risk when combined with pathological mechanotransduction. IOP-independent [e.g., SIX6 (a transcription factor) and CDKN2AS (a non-coding RNA)] genes (12) could compound pressure-mediated injury. Some GWAS loci may be ethnicity-specific: lipid rafts – macromolecular complexes that regulate cell mechanotransduction – have been linked to POAG in European cohorts (23) via mutations in CAV1 and CAV2. Accordingly, cholesterol-lowering drugs (e.g., statins) may decrease the risk of contracting POAG in these populations (24). In addition to changes in gene composition, glaucoma risk may also be associated with copy number variations (base deletions/duplications) (25).
Pressure-dependent dysregulation of signaling mechanisms in anterior eye is reflected in aqueous humor composition, which shows altered levels of a striking number of signaling molecules that include adenosine, nitric oxide, ATP, endothelin-1 (ET-1), lipids (endocannabinoids, prostaglandins, cholesterol), transforming growth factor (TGFβ), CTGF (connective tissue growth factor), angiotensin II and cochlin. ET-1 and TGFβ2 modulate the deposition of ECM molecules within the trabecular pathway, which in turn might affect aqueous outflow (26). Another protein of interest is CYP1B1, a member of the cytochrome P450 enzyme consortium that metabolizes fatty acid molecules downstream from arachidonic acid. CYP1B1 mutations were associated with glaucoma in children and young adults (2), with the protein expressed in glaucoma-relevant ocular tissues such as the trabecular meshwork, retina and the ciliary body where it could affect the cells’ sensitivity to mechanical stressors (27). The calcium-permeable TRPM3 channel (expressed in the ciliary body and lens) represents a combined risk factor for high-tension glaucoma and cataracts (28). Other important signaling mechanisms involve Rho and MAP kinases which modulate the expression of proteolytic enzymes, proinflammatory cytokines, autophagy and contractility (29, 30).
Normal-tension glaucoma (NTG)
NTG is seen in ~30% of POAG patients in which degenerating RGCs show the typical hourglass pattern of axonal loss and visual field defects at “normal” IOP (31), i.e., levels that are consistently lower than 21 mm Hg. Risk factors including female gender, race (more common in Japanese than Caucasians), vascular dysregulation and low blood pressure (32). Two NTG genes (OPTN and TBK1) with prevalence of 1-2% are shared with POAG and encode proteins associated with autophagy (an intracellular protein degradation mechanism) (33). Axonal injury in NTG could be augmented by the pressure gradient caused by abnormally low cerebrospinal fluid pressure across the optic nerve head (32). IOP-lowering regimens improve the prognosis in some patients: after 6 years, ~55% of patients treated with IOP-lowering medications showed stable vision as opposed to ~40% of untreated patients (31, 34). Given that pathological responses NTG patients appear to involve innocuous mechanical stressors, these patients could possess gain-of-function mutations that increase their sensitivity to IOP. Similar hypersensitivity (hyperalgesia) to mechanical stressors is a well-known property of neuropathic pain mechanisms that involves altered function of mechanosensitive ion channels in the peripheral nervous system (35).
Primary Angle-closure glaucoma (PACG)
Angle-closure glaucoma, common in East Asian populations and rare in Caucasians, is responsible for a disproportionate number of patients with severe vision loss. The pathology is caused by disorders of iris, lens and retrolenticular structures which impede drainage of aqueous humor by narrowing the angle between the iris and the cornea (Figure 7B). It is more likely in women and persons with extreme hyperopia (farsightedness). In about a third of PACG patients, an abrupt increase in IOP (often above 30 mm Hg) triggers acute symptoms (severe pain and nausea, conjunctival hyperemia, corneal edema and vomiting) that require immediate medical intervention (12). The disease is highly heritable with gene association studies implicating genes that encode polymorphisms in extracellular matrix (ECM) molecules (e.g., MMP, HSP70, NOS3) or ocular development (the Frizzled related MFRP protein, hepatocyte growth factor HGF). Additional candidates identified by GWAS studies include collagen XI (COL11A1), pleckstrin homology domain proteins (PLEKHA7, PLEKHC1; required for the stabilization of adherens junctions), protein glycosylation (DPM2), acetylcholine biosynthesis (CHAT), ST18 (suppression of tumorigenicity 18) and transcriptional regulation (GLIS3) (36). Unsurprisingly, many of these risk genes encode processes associated with cell adhesion, ECM homeostasis and mechanotransduction (focal adhesion, cell junctions, collagen).
After an acute PACG there may be a period of normal tension, taking the eye multiple days or week to recover from the shock and start producing aqueous fluid at a normal rate that elevates IOP to levels that conform to the extent of angle closure (12).
Secondary glaucoma
Secondary glaucomas reflect pathological mechanisms that elevate IOP above the normal range due to pathological production and/or reduced drainage of aqueous flow. Variants include pseudoexfoliative glaucoma, neovascular glaucoma, pigmentary glaucoma and steroid-induced glaucoma. Pseudoexfoliative glaucoma occurs when clumps of protein accumulate in the anterior chamber and clog the outflow through the trabecular meshwork. It has higher incidence in Scandinavian populations, shares risk factors with Parkinson’s disease and Alzheimer’s diseases and was associated with mutations in the lysyl-oxidase-like 1 (LOXL1) gene, and mutations in loci encoding CACNA1A, TMEM136, SEMA6A and other genes. Neovascular glaucoma: proliferation of blood vessels in the iridocorneal angle (“iris tufts”) results in leakage of fibrous material which closes the angle. Is associated with retinal ischemia. Risk factors include diabetes. Steroid-induced glaucoma occurs in patients who take steroid medications to suppress macular edema, corneal transplant rejection, immune hyper-reactivity and injury-related inflammations. The resulting structural changes in the trabecular meshwork (including the induction of MYOC) increase its resistance to aqueous humor outflow and elevate IOP. Untreated, this induces POAG in 40% of patients (‘steroid responders’) (37). Pigmentary glaucoma typically originates from the “pigment dispersion syndrome” caused by the atrophy of the pigmented epithelial cells in the iris. Dispersal of melanin into the trabecular meshwork impedes aqueous outflow and elevates IOP. The DBA/2J mouse strain, which develops a similar phenotype of chronic glaucoma, has mutations in the Gpnmb gene (encoding a lysosomal-related glycosylated protein) and the Tyrp1 gene (encoding a melanosomal protein with possible enzymatic and structural functions) (38, 39) . Other types of glaucoma are associated with cataract formation, certain eye tumors, uveitis (ocular inflammation), myopia and early onset (juvenile glaucomas).
Aging and glaucoma
Aging and cellular senescence represent a central risk factor for progression of glaucoma. We lose 10,000 ganglion cells (0.3 – 0.6%) and 0.58% of TM cells per year – by the age 80, a healthy individual with normal IOP loses about 30% of their RGCs (8). The loss of ganglion cells in aged individuals correlates with the general decline in cognitive, motor and other functions in aged people and bears many similarities to glaucoma, including dendritic remodeling, reduced axonal transport in the optic nerve (which halves during the lifetime of a rodent), decreased quality control mechanisms in ganglion and trabecular cells, and lower recovery rates following injury. 97.6% of patients over 40 have a median age of glaucoma diagnosis of 64 years, with the prevalence rising steeply from 0.2% – 2.7% in individuals aged 50 – 59 years to 1.6% – 12.8% after age of 80 years (40).
While age is a strong predictor of POAG we don’t know what critical stress is communicated to RGCs in older eyes to make them less resistant to elevated IOP. One possible contributing factor may be the increased stiffness of ocular structures (“ocular rigidity”) which may augment trabecular contractility (which elevates IOP) and increase the susceptibility of ganglion cells and glia to pressure-mediated stress. Older individuals may also have deficient ocular metabolism that is accompanied by age-dependent oxidative stress, mitochondrial dysfunction (41) and loss of neuroprotective mechanisms (42).
Intraocular pressure generation in the eye
It is important to note that IOP reflects the balance between production and outflow of aqueous humor (Figure 7A & Figure 7B). Details of aqueous humor regulation are discussed in reviews by Accott et al. (2014) (43) and Braunger al. (2015) (26) but it is important to note that this transparent, nutritious fluid is continually generated by the ciliary body (~2.4 µl/min at daytime in healthy adults; black arrowhead in Figure 7A) to feed the nonvascularized lens. Aqueous fluid is drained (white arrows) through the trabecular meshwork (TM; red arrowhead, #8 in Figure 7A). In primates, TM supports ~80-90% of aqueous outflow and thus represents the principal bottleneck for fluid outflow whereas the auxiliary ‘uveoscleral’ outflow pathway in the ciliary muscle (#1 in Figure 6A) supports the rest. Aqueous fluid outflow is ~50% lower at night compared with daytime, with consequent increased risk for developing acute pressure-induced eye damage during sleep (44). Both abnormal production of aqueous fluid or its deficient drainage through the TM and the Schlemm’s canal (a pipe composed of endothelial-like cells that feeds the fluid collected from TM into the venous system) can lead to glaucoma.
IOP-dependence contributes a significant proportion of glaucoma cases. Any level of IOP can contribute to pathology but the risk for contracting glaucoma increases exponentially when chronic IOP exceeds 20 mm Hg (Figure 8). The magnitude and duration of IOP correlate with disease severity, with larger IOP elevations precipitating both disease onset and speed of progression (AGIS 2000). Similarly, the longer the duration of ocular hypertension, the higher the likelihood of developing optic neuropathy (31). Importantly, many individuals with elevated IOP do not develop glaucoma and up to 50% of patients with glaucoma (“normal tension” patients) may not have statistically elevated IOP (45). These patients probably express protective factors that counterbalance morbidity factors that drive neurodegeneration whereas “normal tension glaucoma” patients probably express gain-of-function transducer mechanisms that make them super-sensitive to mechanical stress.
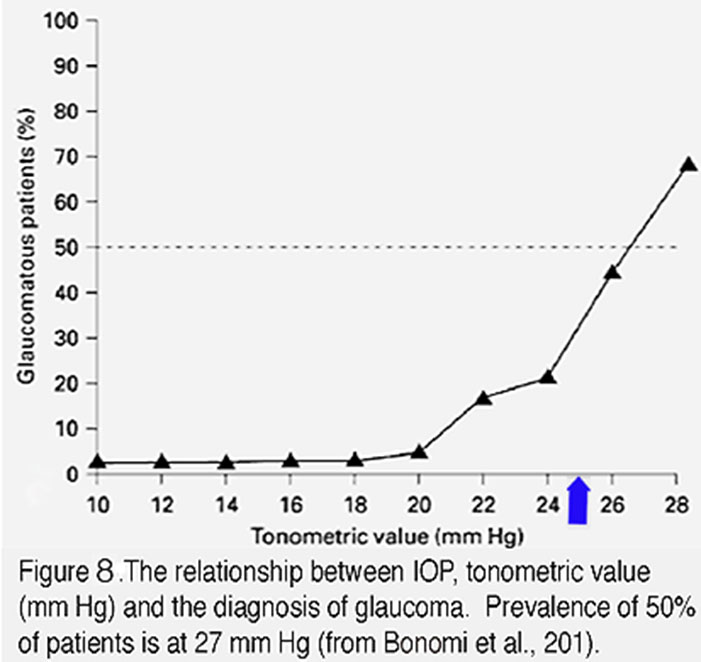 Figure 8. The relationship between IOP, tonometric value (mm Hg) and the diagnosis of glaucoma. Prevalence of 50% of patients is at 27 mm Hg. From Bonomi et al., 2001 (138).
Figure 8. The relationship between IOP, tonometric value (mm Hg) and the diagnosis of glaucoma. Prevalence of 50% of patients is at 27 mm Hg. From Bonomi et al., 2001 (138).
Increased intraocular pressure as a risk factor in glaucoma
The vertebrate eye is a highly dynamic environment where IOP provides the hydrostatic pressure that is necessary for maintaining the shape, growth and optical properties of ocular tissues. Together with old age, increased intraocular pressure (IOP) represents the most important risk factor for the onset and progression of glaucoma (Figure 8). IOP-dependence contributes ~30 – 70% of glaucoma cases (46). The average IOP in newborn rodents and primates that ranges between 6 – 8 mm Hg, increases to 7 – 15 mm Hg in adults. IOP amplitudes tend to be slightly higher in the right eye than the left but the lifetime variability in IOP amplitude for individual eyes is minuscule (<2 mm Hg) (31). In contrast to the average IOP amplitudes which remain stable across lifetime, IOP is complex in the time-domain, showing pulsatility, circadian periodicity and oscillations (~3 mm Hg at ~1 Hz) (47). Blinking, eye scratching, sneezing, lens accommodation, arterial blood pressure and eye movements all transiently elevate IOP (by up to 200 mm Hg). The amplitude and rhythm of IOP fluctuations are regulated by Cry1/Cry2 clock genes and local release of melatonin, which peaks at night and is more pronounced in glaucoma patients (48). IOP and risk for developing glaucoma may be increased by body position (eg, yoga inversion practices), weight lifting and swimming goggles whereas moderate physical exercise and diet rich in ω3/ω6 polyunsaturated fatty acids can decrease IOP by ~4 – 5 mm Hg. A confounding factor, however, is given the wide assortment of risk factors and glaucoma-associated signaling mechanisms there are no clear IOP threshold values that allow us to consistently identify the patients at risk for optic neuropathy.
Currently accepted treatments of glaucoma are largely limited to IOP lowering, a strategy that usually stops disease progression if pressure can be decreased by ~30–50%. Anti-glaucoma medications target aqueous fluid production in the ciliary body and/or the secondary (uveoscleral) outflow pathway. Because IOP lowering may also reduce the progression of vision loss in patients with ‘normal’ IOP (8-15 mm Hg) levels (17), glaucoma can be viewed as a disease that involves increased susceptibility of the eye to mechanical stress at any IOP level. Unfortunately, the most commonly used IOP lowering agent latanoprost (a prostaglandin F2α analog) does not lower IOP more than 20% in a significant fraction (25-50%) of patients whereas up to 6% of POAG patients are unresponsive or do not tolerate any IOP medications (31, 45). Some treatments that showed efficacy in experimental paradigms, such as cannabinoids targeting the CB1 receptor (49), angiotensin II receptor blockers, melatonin receptor 3, adenosine receptor 1 agonists, TRPV4 channel, TREK-1 channel and endothelin receptor-1 blockers have yet to complete clinical trials. No medications were capable of targeting the main (trabecular) outflow mechanism until 2017, when several Rho kinase (ROCK) inhibitors (e.g., ripasudil) were approved for topical treatment of POAG. Unfortunately, the disagreeable side effects (e.g., hyperemia) may limit their usefulness to many patients. Developing drugs that target the trabecular outflow component without side effects should therefore be a priority.
Details of the Trabecular Meshwork (TM)
TM cells play a central role in IOP regulation through their control of fluid outflow into the canal of Schlemm (Figure 7A). The cells form three adjacent strata (uveal, corneoscleral and juxtacanalicular) that differ in terms of their gene expression patterns and relative contribution to phagocytosis, fluid filtration, stiffness and resistance to fluid flow (50) but are difficult to pigeonhole from ontogenetic, morphological or functional standpoints: (i) Similar to macrophages, they express histocompatibility and other immune markers, and show phagocyte-like scavenger activity; (ii) Similar to glia, they play important volume regulatory functions and participate in the release of inflammatory cytokines (such as IL-1α and TNFα); (iii) Similar to endothelia, they form a barrier with antithrombotic and antigen-presenting functions; (iv) Similar to fibroblasts, they release extracellular matrix and contribute to contractility yet (v) They express smooth muscle actin (αSMA), a protein typical of smooth muscle cells. Pressure-evoked TM contractions mediated through actomyosin are thought to elevate IOP by increasing the resistance of the outflow pathway to fluid flow (30). The chronic increase in outflow resistance underlies a positive feedback between elevated IOP, stiffening of the cytoskeleton and increased contractility. In other words, cytoskeletal and ECM remodeling underpin both cytological and functional hallmarks of hypertensive glaucoma. Similar positive feedbacks between mechanical stretch and pathology have been described in other tissues, including lung and heart where cell sensing of small changes in tissue stiffness drives severe, fibrotic remodeling.
At the subcellular level, an increase in pressure induces an upregulation of actin stress fibers, adhesion molecules and remodeling of the extracellular matrix (ECM) in TM cells (Figure 9C & D), together with up/downregulation of hundreds of genes associated with cell adhesion, metabolism, ion transport, inflammation and cell cycle (29). Although not every step of this dynamic response is understood, recent studies showed that mechanical stress induces increases in intracellular calcium concentration [Ca2+]i of both TM cells (Figure 9A) and ciliary body cells that regulate aqueous fluid outflow and inflow, respectively (51). Calcium in turn regulates almost every aspect of cell physiology, including modulation of contractility, stiffness and increased levels of the cytokine TGF-β, activation of Mitogen-Activated Protein (MAP) kinases, release of metalloproteinases (MMPs) and deposition of ECM molecules such as fibronectin and versican (Figure 9D). The effect of pressure on [Ca2+]i and cytoskeletal remodeling that is seen in vitro and in vivo models of trabecular outflow and IOP regulation has been broken experimentally by inhibiting stretch-activated TRPV4 channels (Figure 9B), which triggers an immediate IOP lowering (51).
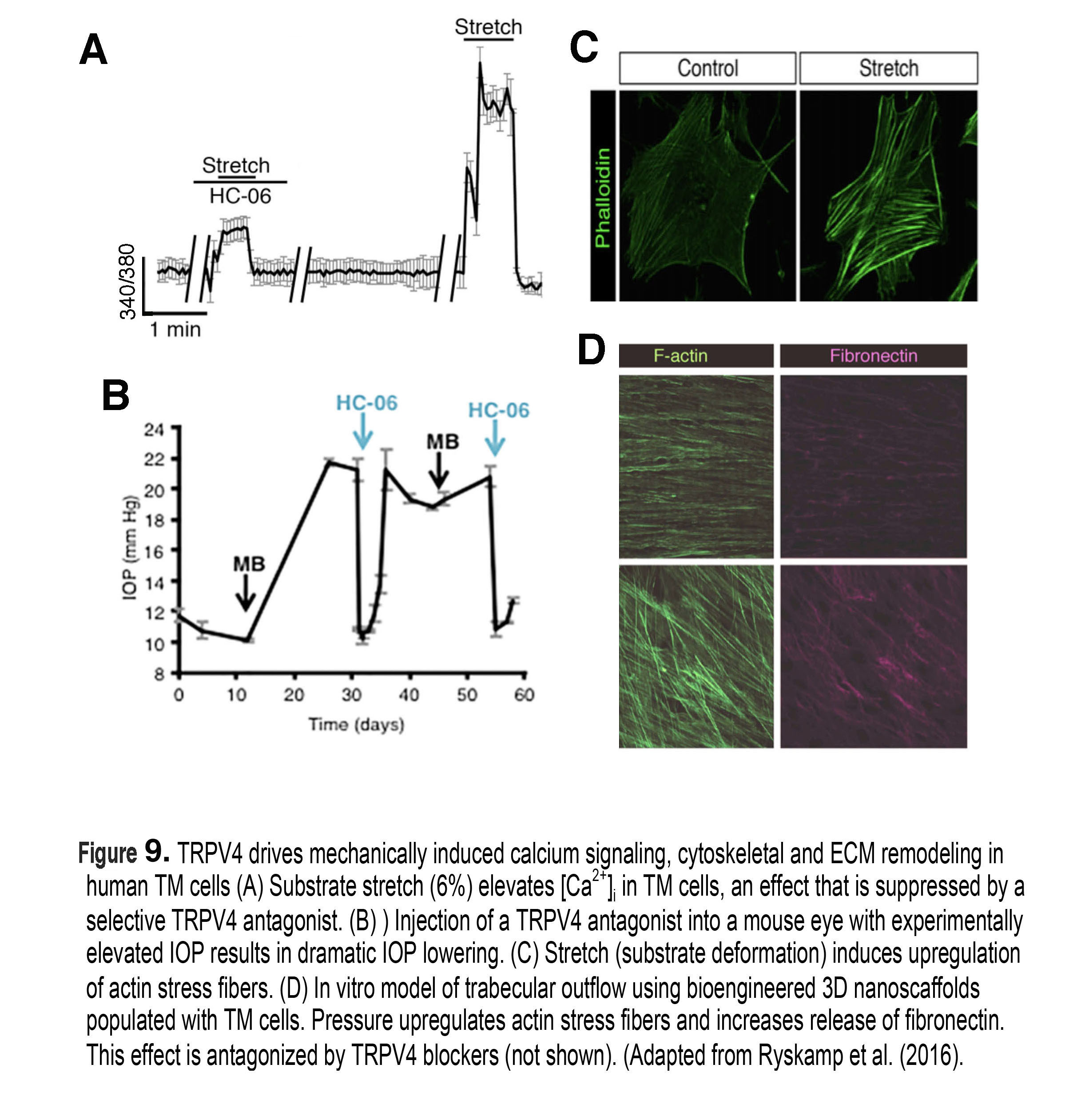 Figure 9. TRPV4 drives mechanically induced calcium signaling, cytoskeletal and ECM remodeling in human TM cells. (A) Substrate stretch (substrate deformation, 6%) elevates [Ca2+)i in TM cells, an effect that is suppressed by a selective TRPV4 antagonist. (B) Injection of a TRPV4 antagonist into a mouse eye with experimentally elevated IOP results in dramatic IOP lowering. (C) Stretch drives the upregulation of ventral actin stress fibers. (D) In vitro model of trabecular outflow using bioengineered 3D nanoscaffolds populated with TM cells. Pressure upregulates actin stress fibers and increases release of fibronectin. This effect is antagonized by TRPV4 blockers (not shown). Adapted from Ryskamp et al (2016) (51).
Figure 9. TRPV4 drives mechanically induced calcium signaling, cytoskeletal and ECM remodeling in human TM cells. (A) Substrate stretch (substrate deformation, 6%) elevates [Ca2+)i in TM cells, an effect that is suppressed by a selective TRPV4 antagonist. (B) Injection of a TRPV4 antagonist into a mouse eye with experimentally elevated IOP results in dramatic IOP lowering. (C) Stretch drives the upregulation of ventral actin stress fibers. (D) In vitro model of trabecular outflow using bioengineered 3D nanoscaffolds populated with TM cells. Pressure upregulates actin stress fibers and increases release of fibronectin. This effect is antagonized by TRPV4 blockers (not shown). Adapted from Ryskamp et al (2016) (51).
Chronic increases in IOP trigger up- and downregulation of thousands of trabecular and retinal genes, many of which encode signaling pathways associated with glial activation, upregulation of complement genes, endothelial dysfunction and RGC degeneration in animals and humans. A useful online resource that showcases the complexity of these pathways is the Glaucoma Discovery Platform from Jackson Laboratories (http://glaucomadb.jax.org/glaucoma).
Details of retinal ganglion cells (RGCs)
Mammals have >45 classes of RGCs – projection neurons with unique inputs, functional properties and axonal projections to the brain (G, red cell in Figure 10). In vitro and in vivo studies in animal models (mainly non-human primates and mice) have established the disproportionate vulnerability of RGCs to many types of mechanical stress – including IOP, compression, tensile stretch and prolonged swelling (35, 52, 53). The spatial pattern of pressure-induced injury of RGCs correlates with the location of their axons within the ONH whereas other classes retinal neurons tend to be (at least initially) spared from IOP-induced injury (Figure 5) (8, 54). The molecular and cellular changes induced by IOP (altered transcription, organelle function, calcium signaling, neurotransmitter uptake, synaptic organization and cellular architecture) in turn compromise the cells’ function that is manifested in their light response, synaptic output and survival. Figuring out why and how IOP preferentially targets RGCs currently represents a central challenge in glaucoma research.
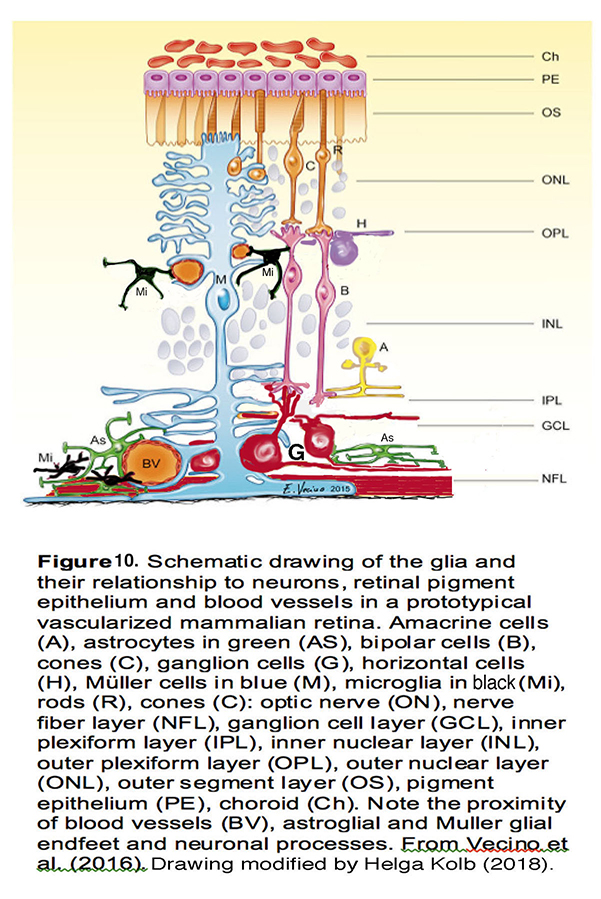
Figure 10. Schematic drawing of the glia and their relationship to neurons, retinal pigment epithelium and blood vessels in a prototypical vascularized mammalian retina. Amacrine cells (A), astrocytes in green (AS), bipolar cells (B), cones (C), ganglion cells (G), horizontal cells (H), Müller cells in blue (M), microglia in black (Mi), rods (R), optic nerve (ON), nerve fiber layer (NFL), ganglion cell layer (GCL), inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), outer nuclear layer (ONL), outer segment layer (OS), pigment epithelium (PE), choroid (Ch). Note the proximity of blood vessels (BV), astroglia, Müller glial endfeet, and neuronal processes. From Vecino et al. (2016) (139). Drawing modified by Helga Kolb. (2018).
{kind=link}
Although ‘glaucoma’ is a late-stage designation of the injured retina, RGCs are remarkably sensitive to acute changes in pressure which result in immediate changes in excitability, loss of spatiotemporal contrast sensitivity and reduced visual acuity (55, 56). Functional dysfunction correlates with the retraction, thinning and reduced complexity of RGC dendrites, loss of synapses and changes in axonal transport (54, 57-60) (Figure 11). Pressure-induced changes in cell firing and dendritic injury are compounded by the obstruction of (anterograde/retrograde) transport of neurotrophic factors and organelles (61) which, if it persists, can contribute to gross structural remodeling at the optic nerve head. Macroscopic remodeling of the ONH tends to be observed months (rodents) or years (primates) after the onset of ocular hypertension, time lags when it tends to be too late for neuroprotective intervention. Recent studies instead suggest that IOP-induced alterations in dendritic architecture and glial activation (see below) represent better early markers of glaucoma than transport loss. The importance of nonaxonal pressure targets was accentuated in a landmark study by Morgan’s group, which demonstrated that pressure-induced dendritic retraction and RGC loss occur in the presence of intact axons (62). The studies of early pressure sensitivity and nonaxonal targets (55, 56) thus emphasize the need for early neuroprotective treatment that targets preservation of dendritic morphology and maintenance of synaptic connectivity within the retina.
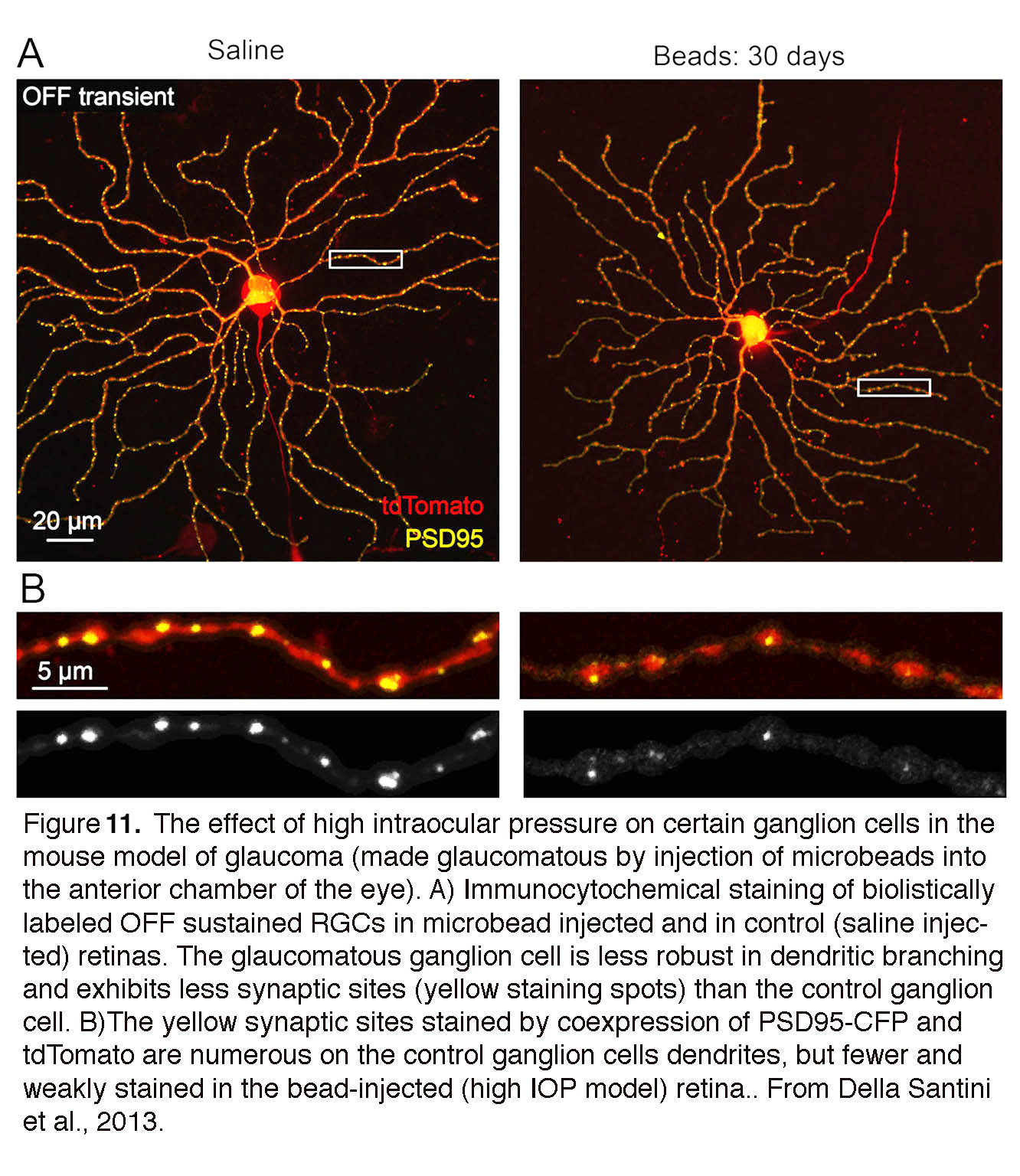 Figure 11. The effect of high intraocular pressure on certain ganglion cells in the mouse model of glaucoma (made glaucomatous by injection of microbeads into the anterior chamber of the eye). (A) immunocytochemical staining of biolistically labeled OFF sustained retinal ganglion cells in microbead injected and in control (saline injected) retinas. The glaucomatous ganglion cell is less robust in dendritic branching and exhibits less synaptic sites (yellow staining spots) than the control ganglion cell. (B) The dendrites are stained by tdTomato, and their synaptic sites by PSD95-CFP. Synaptic sites are more numerous on the control ganglion cell dendrites, but fewer and weakly stained in the bead-injected (high IOP model) retina. From Della Santini et al., 2013 (55).
Figure 11. The effect of high intraocular pressure on certain ganglion cells in the mouse model of glaucoma (made glaucomatous by injection of microbeads into the anterior chamber of the eye). (A) immunocytochemical staining of biolistically labeled OFF sustained retinal ganglion cells in microbead injected and in control (saline injected) retinas. The glaucomatous ganglion cell is less robust in dendritic branching and exhibits less synaptic sites (yellow staining spots) than the control ganglion cell. (B) The dendrites are stained by tdTomato, and their synaptic sites by PSD95-CFP. Synaptic sites are more numerous on the control ganglion cell dendrites, but fewer and weakly stained in the bead-injected (high IOP model) retina. From Della Santini et al., 2013 (55).
There are disagreements regarding the onset of pressure-evoked optic nerve damage and pressure sensitivity of RGC subtypes. An early primate study reported RGCs with large perikarya to be disproportionally sensitive to pressure (63) but these results were not reproduced in primates (64) or rodents, where RGC damage appears to be topographic but not subclass-specific (65). IOP-induced RGC loss was not restricted to specific locations in the retina (55) and there seems to be little correlation between perikaryal size and neurodegeneration (58).
To better understand the disease it would be very helpful to know whether particular classes and subtypes of RGCs are disproportionately sensitive or resistant to mechanical stress. Some electrophysiological recordings combined with analysis of reporter-tagged RGCs point at OFF RGCs as more sensitive to elevated IOP from their ON counterparts (55, 58) but another study, employing similar approaches, reached an opposite conclusion (66). Similarly, Ou et al. (54) found IOP-induced dendritic remodeling to be more extensive in OFF cells yet other studies show no apparent differences in pressure-induced dendritic pruning across functional subtypes (67). It is possible these disparities were caused by different ways of inducing ocular hypertension. In any case, it is very possible that early RGC pressure damage in glaucoma reflects molecular/functional, rather than anatomical, parameters (68). For example, susceptibility of OFF-type cells may reflect different responses from local vasculature or glia, and/or altered availability of trophic factors (58). Intrinsically photosensitive RGCs (ipRGCs) tend to resist IOP elevations (69) (see chapter in Webvision on melanopsin expressing ganglion cells), perhaps due to diminished numbers of pressure transducing molecules (70).
John’s compartmental model (39) transcends such either/or dichotomies by providing a heuristic framework for the early effects of elevated IOP. The model has profoundly advanced the conceptual disease paradigm by positing that RGCs degeneration in glaucoma involves parallel effects of pressure on molecularly distinct local self-destruct programs in dendrites, somata and axons. A plausible multi-compartmental regulator of pressure-induced damage could be the calcium ion, which drives the remodeling of dendrites, somata and axons through Ca2+-dependent proteases, caspases, MAP kinases, ER stress, autophagy and apoptotic cascades (71-73) (see below). Abnormal autophagy and endoplasmic reticulum (ER) damage, which compromise RGC viability due to intracellular accumulation of misfolded proteins, loss of calcium homeostasis and deficient protein trafficking (73) represent additional early features of pressure-related RGC stress, whereas axonal constriction constitutes both a possible early insult and potential coup de grâce that instigates apoptosis. Consistent with the compartmentalization model, local axonal damage can be ameliorated by reducing oxidative damage, targeting the dual leucine zipper kinase (DLK) enzyme and potentially with exercise. Accordingly, preclinical experimental strategies that aim to increase RGC survival in glaucoma can be separated into approaches that (i) facilitate expression of endogenous protective mechanisms, (ii) supply RGCs with exogenous neurotrophic factors, (iii) target pressure-sensing mechanisms, (iv) prevent downstream remodeling (v) slow down cell death by targeting the final common apoptotic pathway and/or (vi) use pharmacological agents with wide-spread or unclear neuroprotective mechanisms of action. Adult ganglion cells in mammals show little or no regenerative response after pressure injury however their survival can be extended for short periods of time with recombinant and virally expressed delivery of neurotrophic factors (e.g., BDNF, CNTF, NT-4, FGF-2, neurturin) which partially protect RGCs in animal models of glaucoma (59, 74). Degrees of protection have been observed following treatments with Rho kinase inhibitors, endothelin ET-2 inhibitors, erythropoietin, resveratrol, NMDA receptor antagonists, NO donors, antioxidants, α2-adrenergic agonists (brimonidine), cannabinoids (e.g., Δ9-THC) and β-adrenergic blockers. Memantine, an antagonist of NMDA receptors, has shielded rodent and non-human primate ganglion cells from glaucoma (75) but was ineffective in human trials. Enigmatically, exposure to long-wavelength red light (850 – 1100 nm) was reported to be protective (76), and visual (neuronal) activity was proposed to facilitate regeneration of injured RGC axons (77). The multiplicity and variability of protective effects suggests that many therapeutic strategies may have been targeting secondary molecular mechanisms that are downstream from the actual RGC mechanotransducers.
Optic nerve head (ONH).
Degeneration (“cupping” or “excavation”) of the ONH is the current diagnostic marker that separates glaucoma from other RGC diseases (e.g., nonarteritic anterior optic neuropathy (NAION), traumatic optic neuropathy, optic neuritis and multiple sclerosis). Cupping correlates with the manifestation of “arcuate scotoma” (loss of sensitivity) in visual testing (perimetry) and correlates with axonal loss observed in imaging studies. Because topology of RGC loss and defects in the visual field (radial in humans, sectorial in rodents) correlate with the position of axons within the ONH (10, 38), the ONH is widely seen to be important in determining the extent of final axonal damage that drives neurodegeneration.
The ONH contains unmyelinated (early) and myelinated (late) parts of RGC axons together with their extracellular matrix ECM/glial scaffolds and the capillary blood supply (Figures 12 & 13) (reviewed in (10, 38)). The biomechanics of this region are affected by the stiffness and thickness of the sclera that surrounds it and by glial deposition of ECM molecules (78). The “papilla” where the optic nerve enters the retina (Figure 12) represents the weakest part of the sclera as the axons (~1.2 million in humans) make a 90o turn to enter the optic nerve through ~200-400 pores of varying sizes that are part of the structure called the lamina cribrosa. 12% of axons must squeeze through these collagenous pores. Axons from the peripheral retina pass in the peripheral part of the lamina whereas those from the central retina pass through its center but axons from the same retinal eccentricity may pass at different levels within the optic nerve.
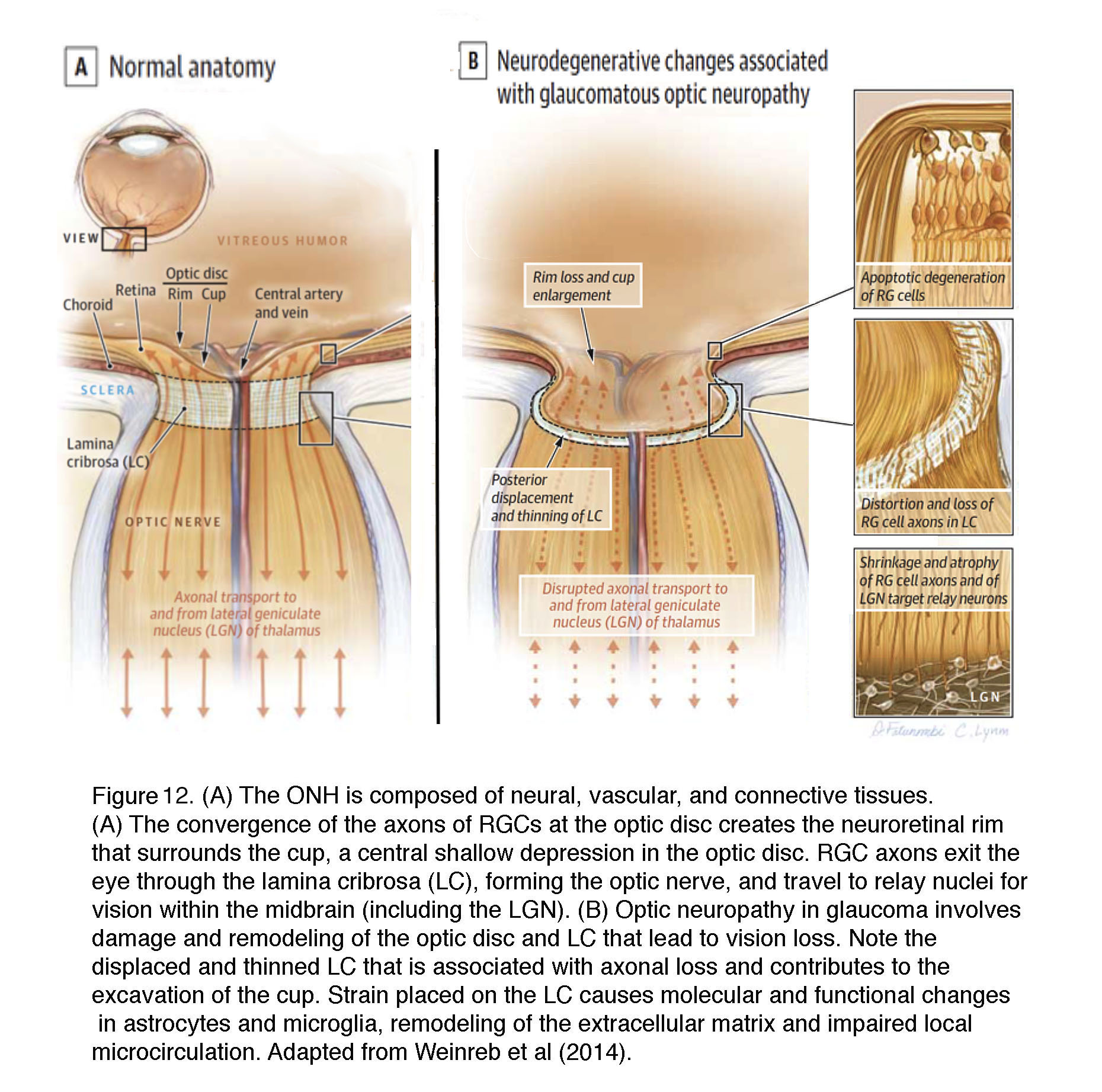 Figure 12. (A) The optic nerve head (optic disk) is composed of neural, vascular, and connective tissues. (A) The convergence of axons of retinal ganglion cells (RG) at the optic disc creates the neuroretinal rim that surrounds the cup, a central shallow depression in the optic disc. RG axons exit the eye through the lamina cribrosa (LC), forming the optic nerve, and travel to relay nuclei for vision within the midbrain (including the LGN). (B) Optic neuropathy in glaucoma involves damage and remodeling of the optic disc and LC that lead to vision loss. Note the displaced and thinned LC that is associated with axonal loss and contributes to the excavation of the cup. Strain placed on the LC causes molecular and functional changes in astrocytes and microglia, remodeling of the extracellular matrix and impairing of local microcirculation. Adapted from Weinreb et al (2014).
Figure 12. (A) The optic nerve head (optic disk) is composed of neural, vascular, and connective tissues. (A) The convergence of axons of retinal ganglion cells (RG) at the optic disc creates the neuroretinal rim that surrounds the cup, a central shallow depression in the optic disc. RG axons exit the eye through the lamina cribrosa (LC), forming the optic nerve, and travel to relay nuclei for vision within the midbrain (including the LGN). (B) Optic neuropathy in glaucoma involves damage and remodeling of the optic disc and LC that lead to vision loss. Note the displaced and thinned LC that is associated with axonal loss and contributes to the excavation of the cup. Strain placed on the LC causes molecular and functional changes in astrocytes and microglia, remodeling of the extracellular matrix and impairing of local microcirculation. Adapted from Weinreb et al (2014).
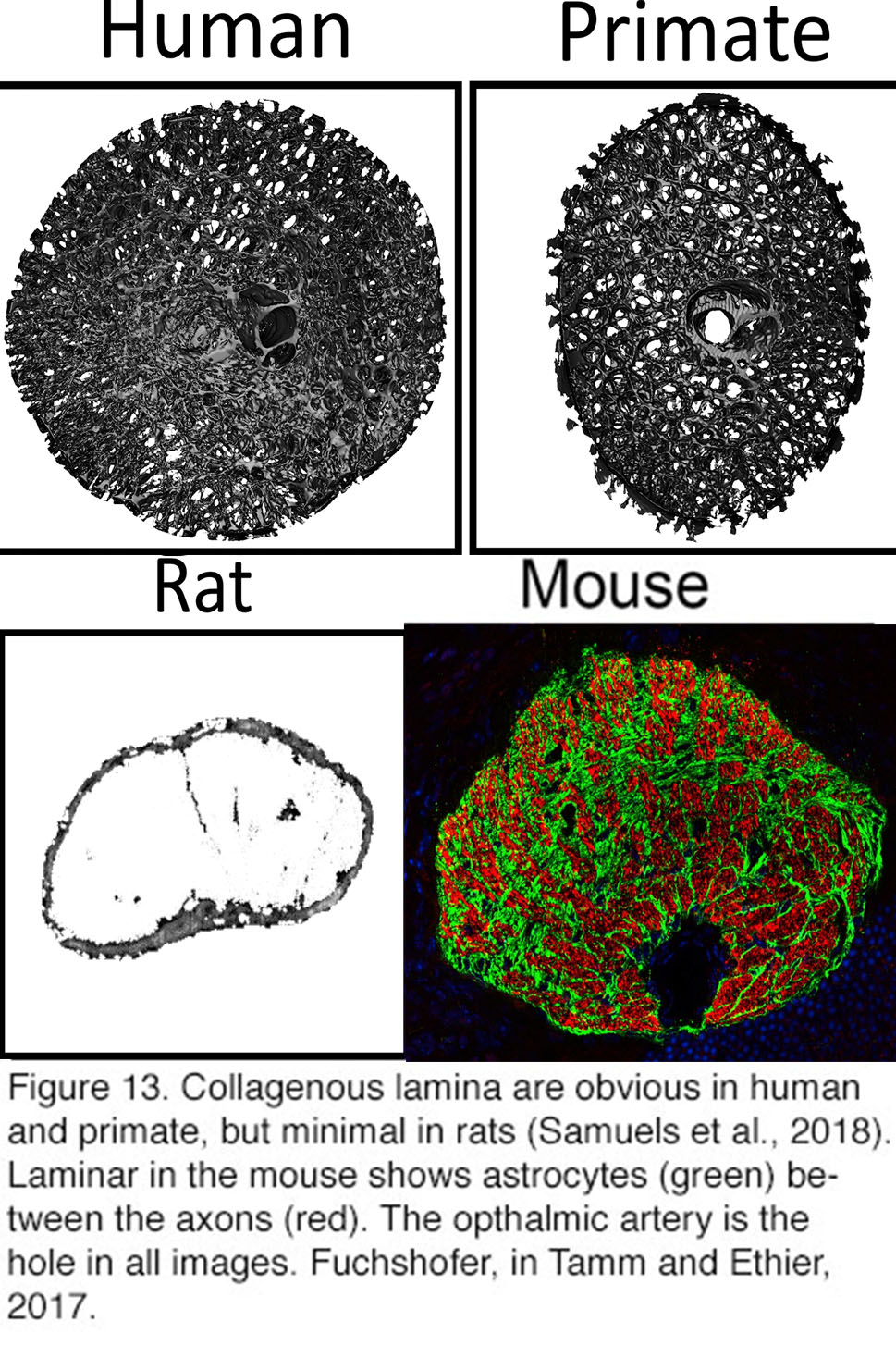 Figure 13. Collagenous lamina are obvious in human and primate, but minimal in rats After Samuels et al., 2018 (140). The lamina in the mouse shows astrocytes (green) between the axons (red). The ophthalmic artery is the hole in all images. From Rudolf Fuchshofer, in Tamm and Ethier, 2017(141).
Figure 13. Collagenous lamina are obvious in human and primate, but minimal in rats After Samuels et al., 2018 (140). The lamina in the mouse shows astrocytes (green) between the axons (red). The ophthalmic artery is the hole in all images. From Rudolf Fuchshofer, in Tamm and Ethier, 2017(141).
The primate lamina cribrosa is a specialized region of ~10 interconnected ECM beams composed of collagen I, III, IV and elastin. These beams are lined with astrocytes, microglia, fibroblasts and an extensively branched vascular supply all of which may interact. Small ECM-cell adhesions may persist for tens of seconds whereas linkages between focal adhesions, intracellular actin cytoskeleton and ECM proteins (fibronectin, tenascin, collagens, etc.) can form on the scales of minutes-to-hours (78). Mechanical strains experienced at the ONH at normal IOPs (~15 mm Hg) range between 5 and 6% whereas IOP elevations to 50 mm Hg may increase them to 8 – 10% (79). Typically, 5-10% cellular stretch is sufficient to substantially affect calcium levels, cytoskeletal organization and tissue physiology across tissues and organs. Biomechanical modeling and 3D reconstruction studies points at the viscoelasticity of ONH structures as a determinant of the dynamic ONH response to changes in IOP. Cribrosal ECM plates in particular are seen as the weakest contact point for mechanical strain/compression (80). Increases in pressure stiffen the ONH yet it is not clear whether a stiffer lamina is better at countering mechanical stress. A useful analogy may be the hypertrophic (fibrotic, stiffer) heart which allows stronger pumping against high back-pressure, but is ultimately detrimental for cardiovascular health.
It must be pointed out that our understanding of the types of strain that affect RGC axons remains incomplete due to the anisotropy of the ONH and the likelihood that subregions with different geometry experience different tensions in response to the same IOP stimulus (78). The size of the pressure differential between IOP and the optic nerve is unknown and it remains unclear that it is affected by cerebrospinal fluid pressure, whether and how stretch of scleral tissues contributes to RGC injury, whether certain RGC classes (which?) are selectively sensitive/resistant to IOP, why retinal glia are so sensitive to elevated IOP, what (if any) is the role of ischemic compression at moderate IOP elevations and which signaling pathways mediate the acute vs. chronic effects of ocular hypertension. These pathways might be linked via IOP-dependent obstruction of anterograde and retrograde transport of pro-survival growth factors (BDNF, NT-3/4, NGF, CNTF), changes in immune gene expression, local chemotaxis, cell-ECM interactions and mitochondria (81, 82). It is possible if not likely that pressure affects these manifold processes via mechanotransducer mechanisms (such as mechanosensitive TRPV4 and TREK-1 channels and integrins; see below), which mediate the responses to scleral stretch, ONH compression, tension & shear. Activation of these mechanotransducers in different cell types (RGCs, glia, vasculature) may determine the relative balance of axonal vs. nonaxonal injury and the importance of neuroinflammation between individuals (and species) with glaucoma.
Many of the studies that implicate axonal involvement in glaucoma have been conducted in optic nerve crush models that are not directly relevant to pressure-induced axonal injury. Studies in animal models of primary open angle (microbead-induced) and secondary closed angle (DBA/2J) glaucoma clearly show that pressure elevations compromise axonal transport (61) but its importance for neurodegeneration remains to be established given that long-lasting reductions in transport don’t impact RGC survival in axonal diseases such as papilledema. Moreover, many types of RGC tolerate loss of transported survival-promoting factors, whereas the lack of sustained RGC survival in retinas supplemented with neurotrophic factors argues against the obligatory consequences of their loss in animals with blocked axonal transport. Furthermore, changes in axonal transport in DBA/2J mice (9-12 months) tend to occur much later (83) than dendritic remodeling (1-3 weeks; (55)), glial reactivity (3-6 months) and defective signaling of the optic nerve (~6 months) (72, 84, 85) . The observation that anterograde transport is compromised before retrograde transport (83) argues against a key role for ONH constriction predicted in most biomechanical models. Instead, recent studies suggest a key role for nonaxonal mechanisms that include physiological deficits in RGC activity (55, 83) and inflammatory signaling (35, 81).
Another important argument against the obligatory role of the lamina cribrosa in glaucoma is supplied by evolution. Laminar ECM plates are well developed in pigs, cats, guinea pigs, tree shrews and dogs but are absent in rodents. In rats and mice, structural and metabolic support to passing RGC axons (~50,000 in mice) is provided by a “glial lamina” composed of astrocytes that form tubes around the axonal bundles (38, 86) (Figure 13). Because these animals still develop glaucoma in response to elevated IOP, neither ECM plates nor the lamina cribrosa are likely to be necessary for pressure-induced axonal loss.
IOP causes neuropathy in the brain
Glaucomatous damage is not restricted to the eye but includes the entire visual pathway from the retina to the brain. IOP increases cause spatial vision loss (87) together with decreases in the size, number and metabolic activity levels in P-, K-and M-layers of the lateral geniculate nucleus (LGN), and cells in magnocellular layers apparently more susceptible to damage compared to parvocellular cells. Furthermore, changes can manifest in layer 4C of the primary visual cortex (88). Hence, regenerative strategies in glaucoma will have to ensure axonal survival and regrowth, perhaps through stimulation of the retinorecipient afferents (89). This will require attention to glaucoma-related changes to RGC axon terminals within the midbrain and retinotopic specificity of ganglion cell projections (61).
Glaucoma effects on the Sclera and Cornea.
Sclera is a dense (400-1000 µm thick) connective-like tissue mainly composed of collagen and elastin fiber and populated with ECM-producing fibroblasts. It extends from the cornea to the optic nerve and provides mechanical support of the retina and an insertion for extraocular muscles. It contacts the lamina cribrosa within the “peripapillary” region (Figure 12A) that is expanded by, and thus resists, IOP. The sclera may therefore be considered biomechanically active system that experiences tension due to IOP and muscular contractions imposed by accommodation/convergence that in turn may be transmitted from the sclera to the adjacent lamina cribrosa, optic nerve and the vasculature that feeds the ONH (78, 80). We know that mechanical stress regulates the composition, growth and size of the eye (90) in part through activity of scleral MMP-2/TIMP2 enzymes (91). Similar to ONH and TM tissues, sclera becomes stiffer and thinner with age and in glaucoma, which may compromise its ability to bear IOP-induced stress and protect the more compliant retina and axons.
Increased risk for IOP-dependent vision loss has been correlated with thinner central corneal thickness and viscoelastic damping (“corneal hysteresis”) (40). This might suggest that glaucoma involves changes in molecular mechanotransduction and biomechanical properties across all tissues in the eye. Subjective vision loss in glaucoma often results from corneal edema caused by a rapid rise in IOP. Unfortunately, little is known about how corneal epithelial/endothelial cells sense pressure and tensile stretch (40).
Neuroinflammation in glaucoma: the crucial functions of glial cells.
Pressure-dependent activation of retinal glia is a central if sometimes underappreciated feature of glaucoma. In healthy retinas, Müller cells (Figure 10, M, blue cells) provide essential metabolic, osmotic support and maintain the excitatory-inhibitory balance through removal of extracellular glutamate. Astrocytes (Figure 10, As, green) maintain the ONH structure, support axons, and maintain the blood-retina barrier. Microglia (Figure 10, Mi, black) regulate proper synapse formation, phagocytosis and immune signaling, whereas oligodendrocytes (not shown in Figure 10) myelinate RGC axons from the lamina to midbrain in the optic nerve.
Glaucomatous glial cells exhibit response patterns that mirror the responsiveness of their brain counterparts to neuronal damage from physical trauma, pathological swelling and neurodegenerative disease. IOP elevations evoke profound changes in retinal glial gene expression, gliotransmitter release, and compromise their roles in nitric oxide, cytokine, chemokine, immune-like molecule (e.g., complement, CX3CL1, TNFα, IL-1β) and reactive oxygen species (ROS) signaling (35, 85). Short and mild increases in IOP may induce reactive activation of Müller cells, astrocytes and microglia prior to significant RGC/axonal damage. One prominent example is glial-dependent changes in cytoskeletal, ECM proteins and structural proteins (the glial acidic fibrillary protein GFAP, vimentin, collagens, laminin and tenascin) and downregulation of proteoglycans (92). Ocular hypertension also underlies metabolomic remodeling in the retinal glia, with includes downregulation of glial glutamate uptake and biosynthesis of glutamine that could impact neuronal excitability and function. A less understood aspect to the yang of pressure-induced proinflammatory glial signaling is the yin of trophic protection from glial release of protective neurotrophic CNTF and/or BDNF factors, and JAK/STAT3 signaling.
Astrocytesin the optic nerve head and in glaucoma
Astrocytes are less abundant in the retina than in the brain but still comprise ~50% of cells in the primate ONH. Their retinal functions include maintenance of vascular tone, ECM production and structural support (93). Astrocytes regulate neuronal activity and neurovascular coupling via ATP, prostaglandin, angiotensin, adenosine and EET (epoxyeicosatrienoic acid) release. Moreover, as the dominant producers of the ECM, astrocytes contribute to the mechanical compliance of the lamina cribrosa and its remodeling in glaucoma. The mammalian retina contains astrocytes with distinct molecular and anatomical signatures. Type I GFAP-positive astrocytes, located in the prelaminar and unmyelinated regions of the human ONH (green in Figures 10 & 14, red in Figure 15A), cover ECM beams and cribrosal pores, ensheath axonal bundles, surround blood vessels and mediate transfer of metabolites between axons and capillaries (green in Figures 10 & 14). Another subtype, the GFAP-negative and α-SMA (smooth muscle actin)- negative Type 2 astrocytes dominate the myelinated postlaminar region of the ONH (94) where they coexist with a subset of recently discovered GFAP- α-SMA+ “lamina cribrosa cells” that may weaken the ONH through dysfunctional synthesis of collagens, elastin and fibronectin (95). In rodents, astrocytes are classified using morphological rather than molecular criteria into protoplasmic and fibrous subtypes (92). As in mammals, rodent astrocytes tile the retina to regulate neurovascular function (Figure 10, Figure15B panel E) and send out fine processes that ensheath and compartmentalize axonal bundles. In contrast to the ONH, rodent astrocytes also provide structural support within the “glial lamina” (Figure 13). However, in animals with avascular retinas (such as rabbits), astrocytes are absent from areas that lack blood vessels.
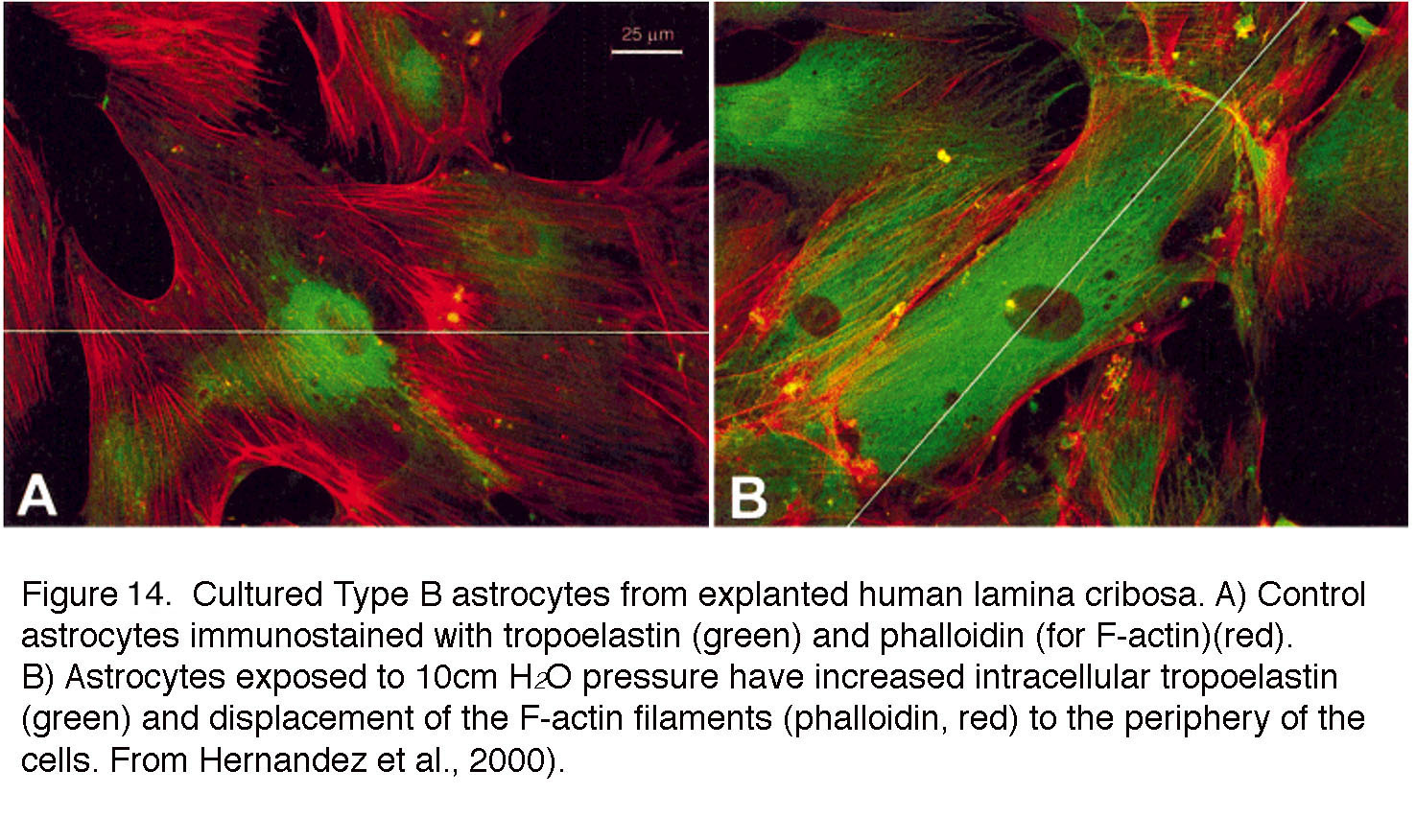 Figure 14. Cultured Type B astrocytes from explanted human lamina cribrosa. (A) Control astrocytes immunostained with tropoelastin (green) and phalloidin (for F-actin, red). (B) Astrocytes exposed to 10 cm H2O pressure have increased intracellular tropoelastin (green) and displacement of the F-actin filaments (phalloidin, red) to the periphery of the cells. From Hernandez et al., 2000 (98).
Figure 14. Cultured Type B astrocytes from explanted human lamina cribrosa. (A) Control astrocytes immunostained with tropoelastin (green) and phalloidin (for F-actin, red). (B) Astrocytes exposed to 10 cm H2O pressure have increased intracellular tropoelastin (green) and displacement of the F-actin filaments (phalloidin, red) to the periphery of the cells. From Hernandez et al., 2000 (98).
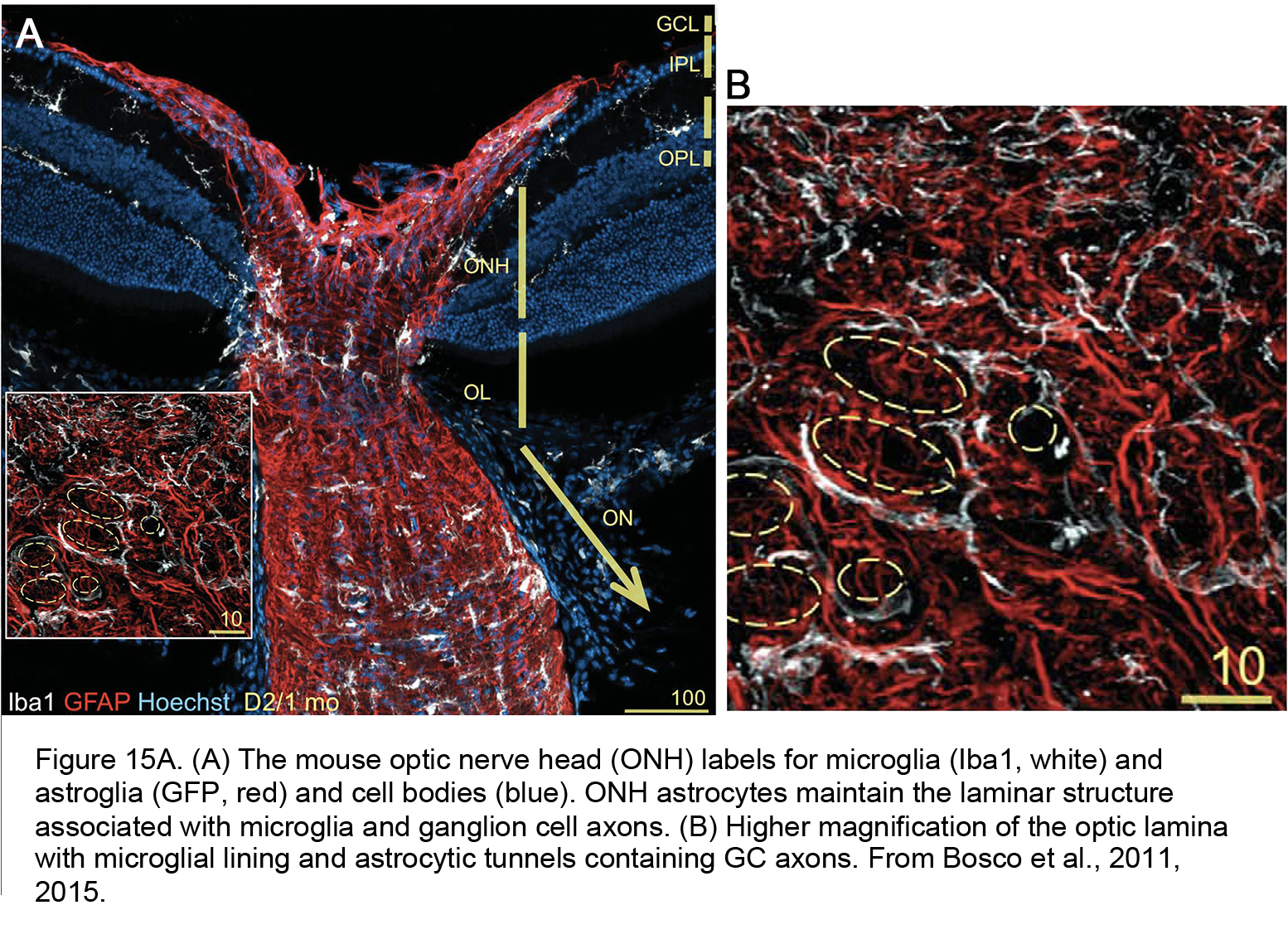 Figure 15A. (A) The mouse optic nerve head (ONH) labels for microglia (Iba1, white) and astroglia (GFP, red) and cell bodies (blue). ONH astrocytes maintain the laminar structure associated with microglia and ganglion cell axons. (B) Higher magnification of the optic lamina with microglial lining and astrocytic tunnels containing GC axons. From Bosco et al., 2011 (84).
Figure 15A. (A) The mouse optic nerve head (ONH) labels for microglia (Iba1, white) and astroglia (GFP, red) and cell bodies (blue). ONH astrocytes maintain the laminar structure associated with microglia and ganglion cell axons. (B) Higher magnification of the optic lamina with microglial lining and astrocytic tunnels containing GC axons. From Bosco et al., 2011 (84).
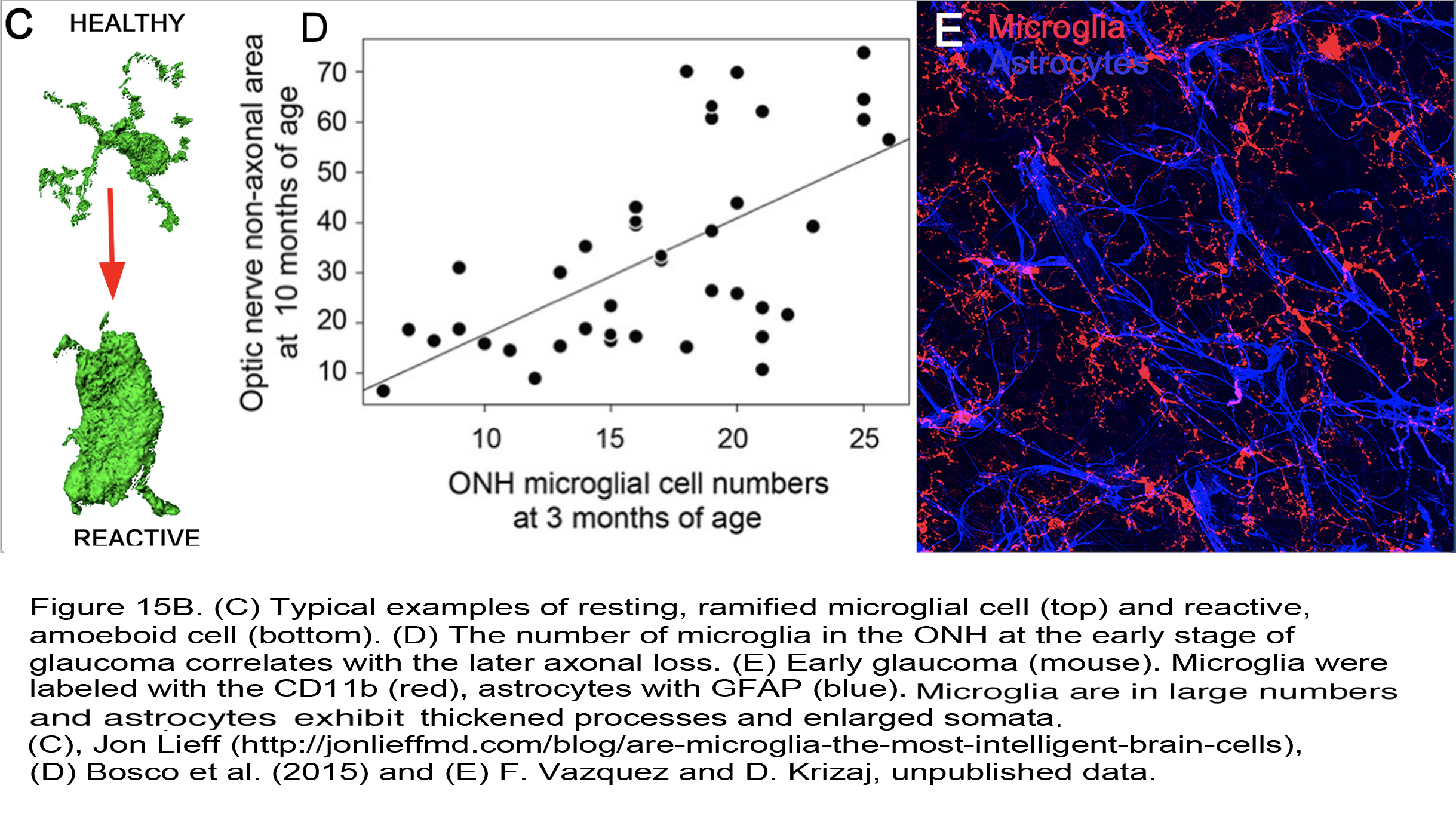 Figure 15B. (C) Typical examples of resting, ramified microglial cell (top) and reactive, amoeboid cell (bottom). (D) The number of microglia in the ONH at the early stage of glaucoma correlates with the later axonal loss. (E) Early glaucoma (mouse). Microglia were labeled with the CD11b (red), astrocytes with GFAP (blue). The cells show signs of hypertrophy (thickened processes and enlarged somata). Images from Jonn Lieff (142) (C), Bosco et al. (2015) (109) (D), and F. Vazquez and D. Krizaj, unpublished data (E).
Figure 15B. (C) Typical examples of resting, ramified microglial cell (top) and reactive, amoeboid cell (bottom). (D) The number of microglia in the ONH at the early stage of glaucoma correlates with the later axonal loss. (E) Early glaucoma (mouse). Microglia were labeled with the CD11b (red), astrocytes with GFAP (blue). The cells show signs of hypertrophy (thickened processes and enlarged somata). Images from Jonn Lieff (142) (C), Bosco et al. (2015) (109) (D), and F. Vazquez and D. Krizaj, unpublished data (E).
Neighboring astrocytes are coupled through homomeric (e.g., Cx 43) and heterotypic gap junctions that redistribute calcium ions and metabolites across the glial network but also regulate neuronal function via release of neuroactive compounds (96, 97). The cells are a major source of fibronectin, but also upregulate many ECM proteins in response to mechanical stressors (pressure, stretch) under both in vivo and in vitro conditions (e.g., tenascin, laminin, collagens, elastin and thrombospondin) (e.g., Figure 14) together with genes associated with cell motility and migration (gelsolin, LIMBP1, periplakin) and downregulation of integrin genes (92, 98).
Mechanical stressors dramatically alter the gene expression, morphology and function of retinal astrocytes and push the cells into a reactive state called astrogliosis that manifests as the inflammation and fibrosis of the lamina, and correlates with weakened ONH architecture, compromised blood supply and axonal loss (86, 98, 99). Astrocytes in the glaucomatous ONH exhibit thickened processes, calcium-dependent hypertrophy and proliferation (86, 99). The list of genes/proteins associated with IOP-induced astrogliosis comprises hundreds of genes that encode cell cycle proteins, mitogen-activated protein kinases (MAPKs), bone morphogenetic proteins (BMPs), endothelin receptors (ETA and ETB), matrix metalloproteinases (MMPs), JAK/STAT3 transcription, cytoskeletal proteins (GFAP, vimentin, collagens, laminin and tenascin), cell adhesion components, signal transduction molecules (NF-kB, STAT5, MCP-1, glypicans), immune/complement systems (C1q, cytokines such as IL-6, LIF), periostin, phagocytosis-related gene Mac-2, and/or NF-κB-dependent assembly of the astroglial inflammasome (81, 100, 101). The cytokine TGF-β2 released from retinal astroglia suppresses MMP-dependent ECM degradation while promoting the expression of collagens, elastin and fibronectin (92) that may result in a stiffer lamina and ONH damage. Another hallmark of pressure-activated astrocytes is the migratory response that allows them to crawl out of laminar cribriform plates (102).
While we don’t understand how elevated IOP induces astrogliosis, physiological studies show a direct link between mechanical stress and astroglial [Ca2+]i (97). Physical forces induce transmembrane currents across retinal astrocyte membranes (103), trigger propagation of calcium waves between astrocytes and Müller glia and stimulate release of proinflammatory mediators. Retinal astroglial mechanotransduction is therefore best viewed within the context of feedback loops with adjacent microglia, Müller cells, microvasculature and RGCs (104).
Microglia and monocytes in glaucoma
Glaucoma involves both adaptive and innate immune mechanisms, with retinal microglia – which constitute the innate immune system – as one of the earliest sentinels of mechanical stress. In healthy retinas, microglial processes (Mi; black in Figure 10) engage in dynamic surveillance of the local milieu and shaping of local synaptic contacts but this activity is dramatically altered in the proinflammatory phenotype associated with glaucomatous microglia. These cells can be characterized by proliferation, process retraction and upregulation of complement, toll-like receptor (TLR2-4), TNFR1α & β receptors, nitric oxide synthase, heat-shock proteins (HSP27, HSP60, HSP72), chemokine (IL-8) and cytokine (IL-1β, IL-6, CCL2, TNFα) signaling as well as the suppression of anti-inflammatory TGFβ signaling (35, 105). Release of metalloproteinases (MMP-1-3) may alter and destabilize the ECM early in the disease. An important recent study from the Vetter laboratory recently demonstrated a key role for microglia in selecting the surviving RGCs during development (106). Within the ONH, microglial activation is first detected in the areas of early axonal damage (Figure 15A, Figure 15B), potentially in response to loss of fractalkine (CX3CL1) inputs from RGCs (107) and altered cross-talk with Müller glia (108). Both minocycline, a small-molecule inhibitor of microglial activation, and bosentan, an inhibitor of microglial endothelin (ET1/2) receptors, increased ganglion cell survival in the DBA/2J mouse glaucoma model (81, 109).
Another important feature of immune activation in the glaucomatous retina is the permeability loss of the blood-retina barrier, which allows the invasion of activated monocytes and dendritic cells from systemic circulation (81, 105). Glaucoma has been linked to microglial changes in the classical and alternative complement pathways, with the ‘alternative’ C1q complex (38) presumably promoting activation of the ‘classical’ C3 convertase that instigates synaptic pruning and somatic phagocytosis. Another important player are the cytokines IL-1β and TNFα, which promote RGC apoptosis in the glaucomatous retina (105). IOP elevation in one eye triggers microglial activation in both eyes, providing evidence that the involvement of immune mechanisms in glaucoma involves the spread of diffusible systemic factors. Consistent with this, activated microglia form clusters around blood vessels in hypertensive eyes (110) and dendritic and synaptic architecture in rodent glaucoma can be protected not only by inhibiting the C1q protein or deleting its gene, but also by blocking pressure-induced immigration of macrophages/monocytes (60, 81). In addition, microglia may respond to mechanically released chemokines such as the monocyte chemoattractant protein (MCP-1) from Müller glia (111) and fractalkine (CX3CL1) from ganglion cells (107).
Müller cells
Müller cells (M; blue in Figure 10) are radial glia that extend from the outer to the innermost retina. They ensheath every neuron and regulate virtually every aspect of retinal function, from energy metabolism to volume regulation, blood flow, uptake and recycling of neurotransmitters and immune signaling (112). Their membrane properties are dominated by potassium channel Kir 4.1-dependent inward rectification, which drowns the contributions from other ion channels. Müller endfeet contribute to the neurogliovascular unit that maintains the blood-retina barrier (Figure 16, control) and also secrete the basement membrane forming the inner limiting membrane through which the retina interacts with IOP. Endfeet strongly express stretch-activated TRPV4 channels and are therefore very sensitive to tensile stretch, poking and changes in volume (27, 113).
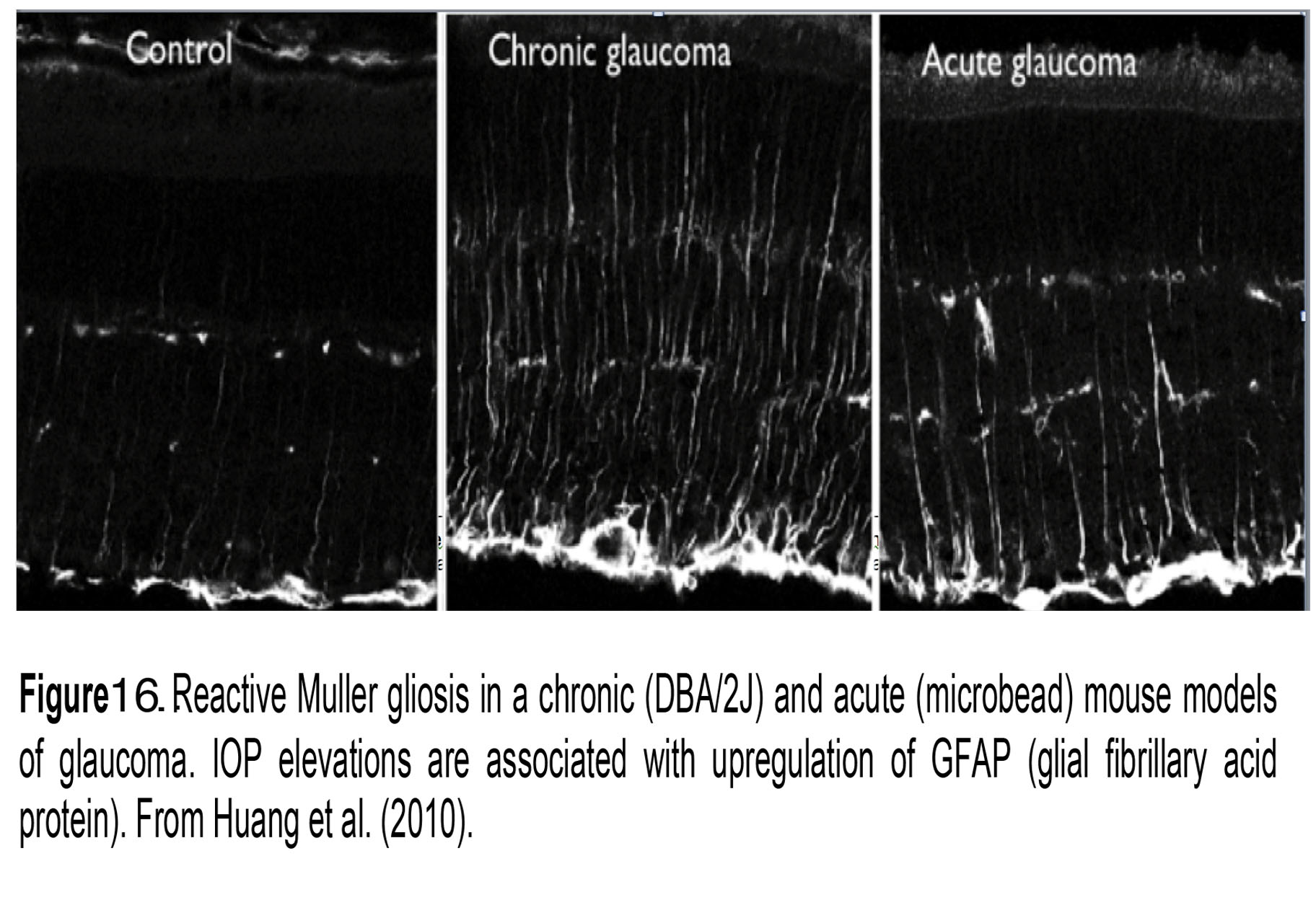 Figure 16. Reactive Müller gloss in a chronic (DBA/2J) and acute (microbead) mouse models of glaucoma. IOP elevations are associated with upregulation of GFAP (glial fibrillary acid protein). From Huang et al. (2010).
Figure 16. Reactive Müller gloss in a chronic (DBA/2J) and acute (microbead) mouse models of glaucoma. IOP elevations are associated with upregulation of GFAP (glial fibrillary acid protein). From Huang et al. (2010).
Müller glia are amongst the first retinal cells to respond to elevated IOP (85). Acute and chronic IOP elevations push them into a reactive state (reactive gliosis) that is typically visualized by immunolabeling the retina for the intermediate filament GFAP (Figure 16; chronic and acute glaucoma). Mice lacking GFAP may be more sensitive to mechanical injury (114), perhaps because these cytoskeletal elements resist mechanical stress. In the reactive mode, Müller cells are likely to modulate vascular permeability and neuronal survival via the release of nitric oxide, TNFα/IL-1β/NF-κB/mTOR signaling and release of the cytokine MCP-1 (35, 111). Among other aspects of Müller signaling affected by pressure are the downregulation of glutamine synthesis/glutamate uptake, upregulation of cytoskeletal proteins, and changes in expression of aquaporin AQP4 (water) channels, calcium channels (ryanodine receptors), complement proteins (C1q) and advanced glycation end products (RAGE) (72, 115, 116).
Endothelial cells, pericytes and local blood supply
Blood supply to the ONH originates in the ophthalmic artery which branches into the central retinal artery, perfusing the nerve fiber layer (Figure 10) and two posterior ciliary arteries. Venous blood drains into the central vein that travels in the optic nerve (Figure 12). Large IOP increases induce ischemia, which compromises the diffusion of glucose, oxygen and lactate through capillary compression, however, even small (3-5 mm Hg) elevations in IOP may result in hypoxia, accumulation of reactive oxygen species and hypoxia-induced factors (e.g., HIF-1α and HIF-2α), downregulation of astroglial glutathione content and induction of the autophagosome (117).
Research challenge for the future
The importance of calcium
The past decades have seen intense efforts to determine the final common mechanism(s) in glaucomatous retinal injury. With respect to RGCs, these mechanisms include ER stress, activation of proapoptotic cascades and dysregulation of calcium homeostasis within axonal and nonaxonal compartments (35, 39). Calcium, a 2nd messenger, plays critical functions in cellular physiology and pathology across the body and it is not surprising that virtually every cell type associated with glaucoma, and every stage of the disease, appear to involve Ca2+ dysregulation. The sources and pathways associated with pathological Ca2+ homeostasis in glaucoma may include ion channels within the plasma membrane and calcium release from intracellular compartments such as the endoplasmic reticulum (ER), mitochondria and lysosomes. It is not inconceivable that such changes in [Ca2+]i promote TM contractility, drive inflammatory activation of retinal glia and facilitate the susceptibility of RGCs to secondary insults.
Experiments in vitro have demonstrated that mechanical stress triggers [Ca2+]i elevations in retinal ganglion cells, microvascular endothelial cells, nonpigmented cells of the ciliary body, glia and the trabecular meshwork (51, 113, 118-120) whereas Ca2+ removal from the external medium or calcium chelation block stretch- and swelling-induced Ca2+ influx and stretch-induced remodeling of the cytoskeleton. There is evidence that [Ca2+]i increases in glaucomatous RGCs occur in dendrites and somata (121), which might account for the increased ER stress and Ca2+-dependent signals (immediate early genes (c-fos, JNK2/3, CREB), proteases, caspases and MAP kinases) in glaucomatous RGCs. Another Ca2+-dependent mechanism involves constitutive activation of calpains, Ca2+-dependent enzymes that promote caspase activation and protein degradation. Inhibition of calpains was protective in models of rodent ganglion cell injury, presumably through the Ca2+-activated phosphatase calcineurin (71). It is also possible, however, that the calcineurin-calpain pathway may contribute to axonal regeneration (122).
The findings described above suggest that targeting pressure-induced calcium influx might help alleviate retinal pathology. Such protective effects were seen under non-physiological conditions (optic nerve transection/crush), with the blockade of voltage- and glutamate-dependent calcium influx conferring largely protective effects on RGC survival and axonal degeneration. We hypothesize that blocking calcium-permeable channels that are sensitive to pressure or its biomechanical corollaries (such as tensile stretch) might result in even more significant injury amelioration in hypertensive eyes.
Importance of mechanotransduction
A crucial requirement without which glaucoma in animals and humans cannot be understood, is to define the molecular links between ocular pressure sensing, aqueous humor dynamics and neurodegeneration. To achieve this, we need a better understanding of actual sites of mechanical impact within the eye and identification of mechanical stressors (tensile strain, shear flow, compression etc.) that are sensed by cells in both the anterior and the posterior eye. Studies in animal models of glaucoma and in isolated cells show that most glaucoma-relevant cell types (trabecular meshwork cells, retinal glia and RGCs) respond to pressure with fast onset electrical and calcium signals (~milliseconds to ~seconds), whereas sustained exposure to mechanical stressors tends to evoke phenotypes that are characteristic of glaucomatous injury. In addition to characterizing the pressure transducer molecules, it will also be important to determine how ocular cells ignore steady-state IOP, and how they respond to and compensate for transient IOP fluctuations triggered by blinking, coughing, eye rubbing, circadian periodicity, swelling, ocular trauma, and exercise (e.g., swimming goggles, yoga practice). To identify the molecular mechanisms, we can now build on the vast trove of information available from investigations of cellular mechanosensing.
Mechanobiology is an emerging field that combines biology and bioengineering to study how physical forces and changes in mechanical properties of cells contribute to cell development, differentiation, proliferation and disease. Its common theme is that every cell converts mechanical inputs into electrical or biochemical signals by using specialized proteins that govern the mechanical stability and mechanical signal transmission at virtually every level of organization, from the nanoscale to macroscale (123, 124). These mechanotransduction machines impose regulatory constraints at the level of cell-cell and tissue-tissue networks but some cells are better at mechanotransduction than others, and therefore more sensitive to mechanical forces and dysregulated in disease. Often cited examples of mechanotransduction gone awry are hearing and balance diseases caused by mutations in mechanosensitive proteins, but there are many other examples of bone, cartilage, lung, bladder, immune system, heart and eye diseases caused by increased pressure and stretch (125). Glaucoma and myopia, for example, can be considered as mechanotransduction diseases that reflect the dysfunctional response of cells in the eye to specific types of local mechanical stress.
Many protein families respond to mechanical forces with conformational changes. Mechanosensitive proteins include focal adhesion proteins, cell-cell adhesion, integrin and ECM proteins, receptor tyrosine kinases, G-protein-coupled receptors, cytoskeletal proteins and, importantly, ion channels that are activated by minute changes in membrane curvature and stretch in the absence of chemical ligands (124-126). These mechanosensitive channels evolved for sensing submicrometer displacements of the plasma membrane but show huge differences in primary structure, ion selectivity, conductance and pharmacology (Figure. 17). They have been classified into Na+-permeable (ENaC/degenerins), K+-permeable (TREK, TRAAK) and nonselective cation (TRP and piezo) channel families, which show isoform-specific mechanical thresholds, time courses, adaptation to sustained stimulation as well as differences in how they tether to the cytoskeleton and respond to posttranslational modulation.
Conceptually, mechanochannels function as bandpass filters that can transmit specific amplitudes and types of mechanical stress (i.e., compression, tension, swelling, shear flow etc.) that range from gentle touch, pressure, texture, vibration, sound, shear flow to physical trauma and pain. Recent studies show that glaucoma-relevant tissues in the eye (TM, ciliary body, neurons, glia, cornea, endothelial cells) express multiple types of mechanosensitive TRP, piezo and tandem pore K+ channels (27, 127-129). Pharmacological suppression and genetic ablation of these channels ameliorate the inflammatory and degenerative retinal phenotypes associated with ocular hypertension (119), suggesting possible roles in glaucoma.
Figure 17 illustrates the basic topology of vanilloid TRP (TRPV) and TREK (TWIK-1 related K+) mechanotransducers. TRPV4, a member of the vanilloid TRP family mediates influx of Na+ and Ca2+ ions in response to a wide variety of mechanical and chemical stimuli (130, 131) that include pressure, stretch, swelling, poking and body temperature (34 – 37oC). It is expressed in ~60-70% of mouse RGCs, 100% of Müller cells (Figure 18A) and the majority of trabecular and corneal epithelial cells (70, 113, 132), with some RGCs sharing TRPV4 expression with the cognate TRPV1 channel (Figure 18B) (70, 128). Interestingly, the channel appears to be absent from neuronal classes that are less sensitive to glaucoma (photoreceptors, bipolar, amacrine cells; Figure 10) and mice lacking TRPV4 show no deficits in outer retinal function (Figure 18C). TRPV4 channels are often integrated into cells’ cytoskeleton network through ankyrin repeats in the N-terminus (Figure 17A) and are linked to ECM networks through α2 integrin molecules and focal adhesions. They are sensitive to pressure (129), drive stretch-induced cytoskeletal remodeling and may contribute to the loss of tensile homeostasis during chronic IOP elevations (51).
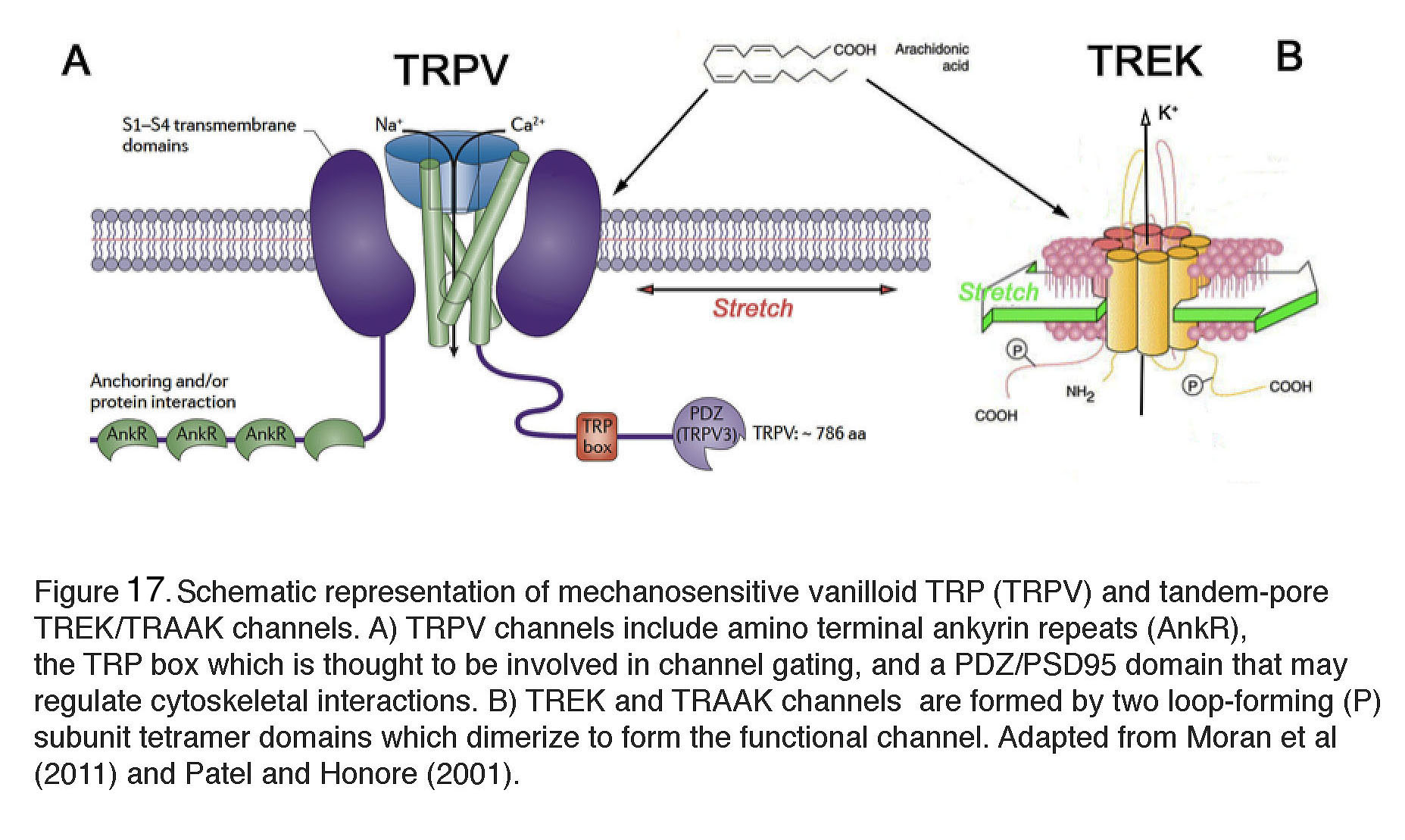 Figure 17. Schematic representation of mechanosensitive vanilloid TRP (TRPV) and tandem-pore TREK/TRAAK channels. (A) TRPV channels include amino terminal ankyrin repeats (AnkR), the TRP box which is thought to be involved in channel gating, and a PDZ/PSD95 domain that may regulate cytoskeletal interactions. (B) TREK and TRAAK channels are formed by two loop-forming (P) subunit tetramer domains which dimerize to form the functional channel. Adapted from Moran et al., 2011 (143) and Patel and Honoré, 2001 (144).
Figure 17. Schematic representation of mechanosensitive vanilloid TRP (TRPV) and tandem-pore TREK/TRAAK channels. (A) TRPV channels include amino terminal ankyrin repeats (AnkR), the TRP box which is thought to be involved in channel gating, and a PDZ/PSD95 domain that may regulate cytoskeletal interactions. (B) TREK and TRAAK channels are formed by two loop-forming (P) subunit tetramer domains which dimerize to form the functional channel. Adapted from Moran et al., 2011 (143) and Patel and Honoré, 2001 (144).
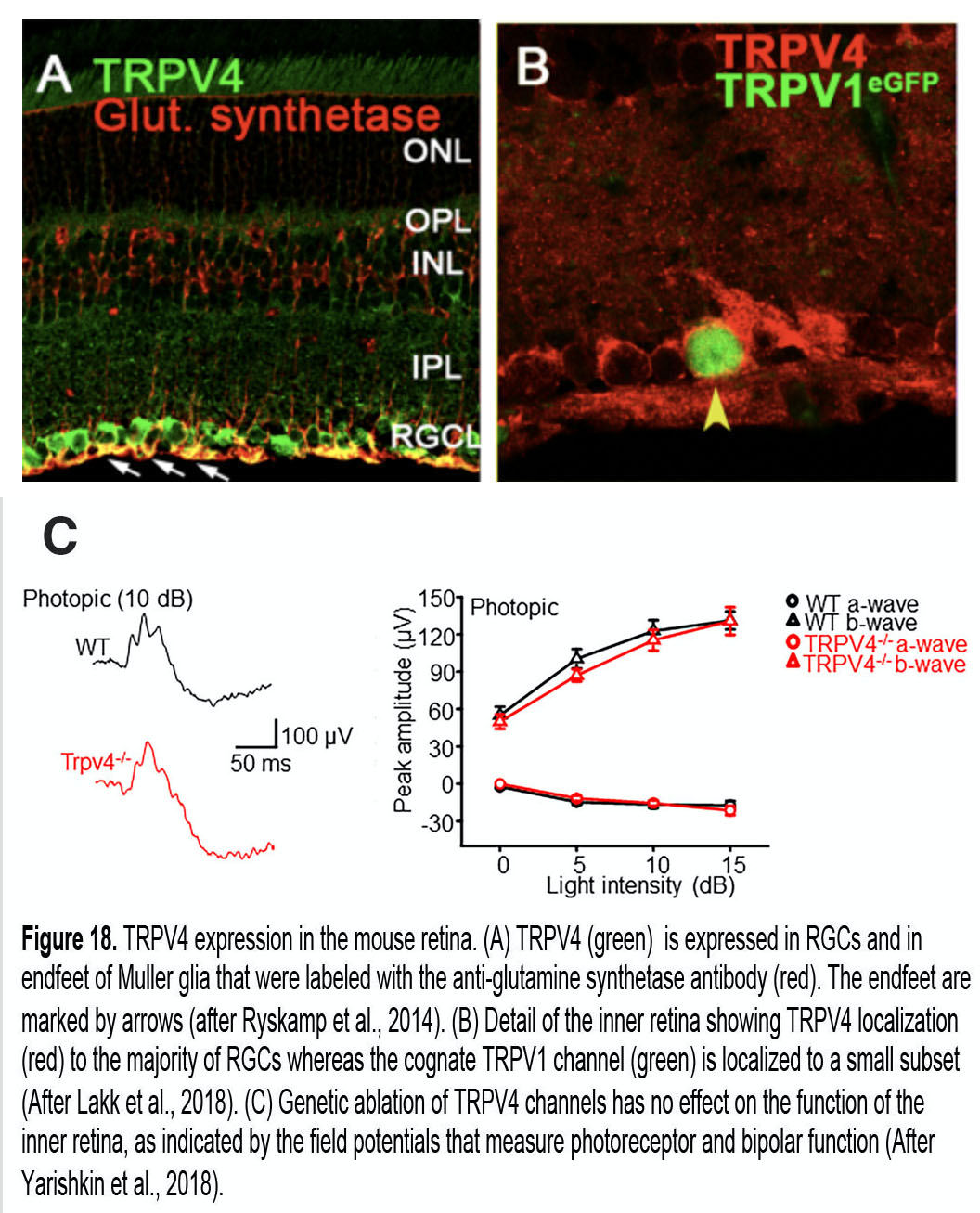 Figure 18. TRPV4 expression in the mouse retina. (A) TRPV4 (green) is expressed in retinal ganglion cells and in the endfeet of Müller glia that were labeled with the anti-glutamine synthetase antibody (red). The endfeet are marked by arrows. After Ryskamp et al., 2014 (27). (B) Detail of the inner retina showing TRPV4 localization (red) to the majority of retinal ganglion cells, whereas the cognate TRPV1 channel (green) is localized to a small subset. After Lakk et al., 2018 (70). (C) Genetic ablation of TRPV4 channels has no effect on the function of the inner retina, as indicated by the field potentials that measure photoreceptor and bipolar function. After Yarishkin et al., 2018 (145).
Figure 18. TRPV4 expression in the mouse retina. (A) TRPV4 (green) is expressed in retinal ganglion cells and in the endfeet of Müller glia that were labeled with the anti-glutamine synthetase antibody (red). The endfeet are marked by arrows. After Ryskamp et al., 2014 (27). (B) Detail of the inner retina showing TRPV4 localization (red) to the majority of retinal ganglion cells, whereas the cognate TRPV1 channel (green) is localized to a small subset. After Lakk et al., 2018 (70). (C) Genetic ablation of TRPV4 channels has no effect on the function of the inner retina, as indicated by the field potentials that measure photoreceptor and bipolar function. After Yarishkin et al., 2018 (145).
Many ocular manifestations of glaucoma (IOP changes, calcium overloads, reactive gliosis, cytokine release, microvascular dysfunction, actin upregulation, fibronectin secretion and RGC apoptosis) can be mimicked pharmacologically by stimulating TRPV4 channels in RGCs, glial, endothelial, ciliary body and trabecular meshwork cells (27, 111, 127, 133). TRPV4-induced phenotypes include RGC degeneration (133), reactive gliosis (27), glial swelling (113) and increased permeability of the blood-retina barrier (127, 134) whereas mice lacking the TRPV4 gene seem to be protected from some types of mechanical and nociceptive stress (130) and do not appear to develop glaucoma (51). Cholesterol and polyunsaturated fatty acids – potential risk factors in glaucoma – also modulate TRPV4 activation (135).
How is mechanical homeostasis in the healthy eye maintained in counterpoint to TRP and piezo channel activation? A possible mechanotransducer candidate that opposes excessive TRPV4 activation might be the K+-permeable TREK-1 channel. TREK-1 is eerily sensitive to stimuli that gate TRPV4 (membrane stretch, swelling, body temperature, and arachidonic acid) and is often co-expressed with TRPV4 (O. Yarishkin, F. Vazquez-Chona and D.K., unpublished observations). Mice lacking TRPV4 channels are less sensitive to mechanical stress (131) whereas mice with ablated TREK/TRAAK channels are hypersensitive to mechanical forces and local pH changes (136). According to this model, the tensile homeostasis of at least some ocular tissues might be maintained by concomitant activation of TRPV4/piezo vs. TREK-1 (and its cognate TRAAK) channels.
Summary and Conclusion
Glaucoma is a leading cause of irreversible blindness worldwide. There are multiple forms of the disease, but all involve dysfunctional signaling in the anterior eye that is linked to optic neuropathy. There is no cure and at present there is no way to regain sight that had been lost. This is a problem, because glaucoma is often asymptomatic until very late stages and many people with the disease do not realize they have the disease until it is too late for effective treatment. It is therefore important that patients with risk factors (i.e., elderly individuals with family history and/or African ancestry) are screened at regular intervals. After diagnosis, treatment will be lifelong but requires vigorous compliance with the treatment regimen (IOP-lowering eye drops).
What is glaucoma? The clearest answer is that we still don’t know, as pathology can reflect a multifactorial pathology and/or a collection of diseases with a similar final common pathology. Interestingly, mechanotransduction in aged individuals and those with glaucoma appear to be compromised across all tissues of the eye (cornea, sclera, TM, retina), suggesting broad dysfunctions in the cells’ ability to generate and process mechanical forces. Most forms of glaucoma appear to be hereditary, but it is likely that in order for the pathology to manifest, mutations/alterations in multiple proteins and/or lipids are required and identification of causal genes and mutations will immensely advance the diagnosis and treatment. For now, only 5-10% of cases have been linked to specific genes.
IOP lowering – currently the main treatment – begin with eye drops, with refractory or advanced cases relegated to laser procedures and incisional surgeries. At this time, anti-glaucoma agents largely target a secondary pathway that mediates a minuscule fraction of fluid outflow and there is much scope for improvement, especially in targeting trabecular outflow. Another priority is to protect retinal neurons and glia from pressure, which necessitates the elucidation of molecular/cellular mechanisms that sense and transduce mechanical stress. Specifically, improved treatment requires research scientists, clinicians and biomedical engineers to (i) Determine the quantitative relationships between risk factors, genetic susceptibility and molecular mechanisms that underlie cellular interactions in the front and the back of the eye, (ii) Develop standardized tests to define where the optic neuropathy in a particular patient falls in the spectrum between ‘normal’ aging and glaucoma, (iii) Develop novel technologies (such as optical coherence tomography) for early diagnosis (iv) Improve the efficacy of pharmacological agents that target TM while eliminating side effects and (v) Protect the pressure sensitive targets within the trabecular meshwork, retinal ganglion cells and glia. This requires investment into neuroprotective treatments, which has been discouraged by disagreements among scientists about RGC mechanotransduction mechanisms, the prohibitive expense of clinical trials, and recent pricey failures of putative neuroprotection drugs.
A main goal for future treatments must be to lower IOP together with the protection from pressure-dependent inflammation and neurodegeneration, an approach that may include direct targeting of the pressure sensing proteins themselves. Unfortunately, at this moment there seems to be – despite decades of intense research – no consensus regarding the early retinal targets of IOP (dendrites? somata? axons? glia? blood vessels? ONH?). It is becoming obvious, for example, that glia are amongst the earliest responders to elevated pressure and that neurodegeneration can continue long after IOP is stabilized. Therefore, any attempts at neuroprotection must take into account IOP-independent vascular, glial and scleral mechanisms. Very possibly, prevention of glaucoma will improve with better understanding of molecular, circadian, caloric restriction and exercise parameters that are increasingly viewed as relevant to ocular health. Another innovative front will definitely be on the technology side, as patients are equipped with real-time telemetric devices, noninvasive ONH measurements become routine, and diagnoses are provided by deep learning (artificial intelligence) algorithms (46). We live in exciting times that, having unveiled the key likely components and patterns of visual damage in glaucoma and old age, can only lead to glorious future where mechanotransduction mechanisms in individuals at risk for disease onset are understood, targeted and used for early diagnosis.
Acknowledgements
Supported by the National Institutes of Health, The Willard L. Eccles Foundation, Glaucoma Research Foundation, University of Utah Technology Acceleration Grant and an Unrestricted Grant from Research to Prevent Blindness to the Department of Ophthalmology at the University of Utah. The author thanks the past and present members of his laboratory for outstanding work over the past decade, Andrea Blitzer, M.D. for help with initial literature review and Drs. Ralph Nelson (NIH) and Helga Kolb (University of Utah) for helpful comments.
About the author

David Krizaj received his B.A. at the University of Ljubljana (Slovenia), Ph.D. in Physiology & Biophysics at New York University and completed postdoctoral studies in Retinal Neurobiology at University of California San Francisco. He took a position at the Moran Eye Center at the University of Utah School of Medicine where he holds the John Frederick Carter Endowed Professorship in Ophthalmology. Dr. Krizaj’s interests include intracellular signaling pathways with a focus on calcium homeostasis, synaptic transmission and mechanotransduction, and the last 5 years his lab has been actively engaged in studying the roles of mechanosensitive TRP, tandem-pore and piezo channels in the regulation of intraocular pressure. Another main thrust is to define the molecular mechanisms of pressure sensitivity of trabecular meshwork cells, retinal neurons and glia, and how its dysregulation contributes to diabetic retinopathy and glaucoma. Dr. Krizaj can be contacted at david.krizaj@hsc.utah.edu.
References
- Flaxman, S.R., R.R. Bourne, S. Resnikoff, P. Ackland, T. Braithwaite, M.V. Cicinelli, A. Das, J.B. Jonas, J. Keeffe, and J.H. Kempen, Global causes of blindness and distance vision impairment 1990–2020: a systematic review and meta-analysis.The Lancet Global Health. 2017; 5(12):e1221-e1234. [PubMed]
- Ko, Y.-C., D.-K. Hwang, W.-T. Chen, C.-C. Lee, and C.J. Liu, Impact of socioeconomic status on the diagnosis of primary open-angle glaucoma and primary angle closure glaucoma: a nationwide population-based study in Taiwan.PloS one. 2016; 11(2):e0149698. [PubMed]
- Tsatsos, M. and D. Broadway, Controversies in the history of glaucoma: is it all a load of old Greek?British Journal of Ophthalmology. 2007; 91(11):1561-1562. [PubMed]
- Leffler, C.T., S.G. Schwartz, F.M. Giliberti, M.T. Young, and D. Bermudez, What was glaucoma called before the 20th century? Ophthalmology and eye diseases. 2015; 7:OED. S32004. [PubMed]
- Mackenzie, W., A. Hewson, and T.W. Jones, A Practical Treatise on the Diseases of the Eye. 1855: Blanchard and Lea. [PubMed]
- Hood, D.C., Improving our understanding, and detection, of glaucomatous damage: an approach based upon optical coherence tomography (OCT).Progress in retinal and eye research. 2017; 57:46-75. [PubMed]
- Smith, C., J. Vianna, and B. Chauhan, Assessing retinal ganglion cell damage.Eye. 2017; 31(2):209. [PubMed]
- Harwerth, R.S., M. Crawford, L.J. Frishman, S. Viswanathan, E.L. Smith III, and L. Carter-Dawson, Visual field defects and neural losses from experimental glaucoma.Progress in retinal and eye research. 2002; 21(1):91-125. [PubMed]
- Pavlidis, M., T. Stupp, R. Naskar, C. Cengiz, and S. Thanos, Retinal ganglion cells resistant to advanced glaucoma: a postmortem study of human retinas with the carbocyanine dye DiI.Investigative ophthalmology & visual science. 2003; 44(12):5196-5205. [PubMed]
- Nickells, R.W., G.R. Howell, I. Soto, and S.W. John, Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy.Annual review of neuroscience. 2012; 35:153-179. [PubMed]
- Libby, R.T., D.B. Gould, M.G. Anderson, and S.W. John, Complex genetics of glaucoma susceptibility.Annual review of genomics and human genetics. 2005; 6. [PubMed]
- Wiggs, J.L. and L.R. Pasquale, Genetics of glaucoma.Human molecular genetics. 2017; 26(R1):R21-R27. [PubMed]
- Jonas, J.B., T. Aung, R.R. Bourne, A.M. Bron, R. Ritch, and S. Panda-Jonas, Glaucoma.Lancet. 2017; 390(10108):2183-2193. [PubMed]
- Quigley, H.A., Understanding glaucomatous optic neuropathy: the synergy between clinical observation and investigation.Annual review of vision science. 2016; 2:235-254. [PubMed]
- Weinreb, R.N., T. Aung, and F.A. Medeiros,The pathophysiology and treatment of glaucoma: a review.Jama. 2014; 311(18):1901-1911. [PubMed]
- Allingham, R.R., K.F. Damji, and M.B. Shields, Shields textbook of glaucoma. 6th ed. 2011, Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. xiv, 610 p.
- Jonas, J.B., P. Weber, N. Nagaoka, and K. Ohno-Matsui, Glaucoma in high myopia and parapapillary delta zone.PLoS One. 2017; 12(4):e0175120. [PubMed]
- Tham, Y.-C., T. Aung, Q. Fan, S.-M. Saw, R.G. Siantar, T.Y. Wong, and C.-Y. Cheng, Joint effects of intraocular pressure and myopia on risk of primary open-angle glaucoma: the singapore epidemiology of eye diseases study.Scientific reports. 2016; 6:19320. [PubMed]
- Armaly, M., The genetic determination of ocular pressure in the normal eye.Archives of Ophthalmology. 1967; 78(2):187-192. [PubMed]
- Khor, C.C., T. Do, H. Jia, M. Nakano, R. George, K. Abu-Amero, R. Duvesh, L.J. Chen, Z. Li, and M.E. Nongpiur, Genome-wide association study identifies five new susceptibility loci for primary angle closure glaucoma.Nature genetics. 2016; 48(5):556. [PubMed]
- Zode, G.S., K.E. Bugge, K. Mohan, S.D. Grozdanic, J.C. Peters, D.R. Koehn, M.G. Anderson, R.H. Kardon, E.M. Stone, and V.C. Sheffield, Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma.Investigative ophthalmology & visual science. 2012; 53(3):1557-1565. [PubMed]
- Minegishi, Y., M. Nakayama, D. Iejima, K. Kawase, and T. Iwata, Significance of optineurin mutations in glaucoma and other diseases.Progress in retinal and eye research. 2016; 55:149-181. [PubMed]
- Thorleifsson, G., G.B. Walters, A.W. Hewitt, G. Masson, A. Helgason, A. DeWan, A. Sigurdsson, A. Jonasdottir, S.A. Gudjonsson, and K.P. Magnusson, Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma.Nature genetics. 2010; 42(10):906. [PubMed]
- McGwin, G., S. McNeal, C. Owsley, C. Girkin, D. Epstein, and P.P. Lee, Statins and Other Cholesterol-Lowering Medications and the Presence of Glaucoma.Archives of ophthalmology. 2004; 122(6):822-826. [PubMed]
- Davis, L.K., K.J. Meyer, E.I. Schindler, J.S. Beck, D.S. Rudd, A.J. Grundstad, T.E. Scheetz, T.A. Braun, J.H. Fingert, and W.L. Alward, Copy number variations and primary open-angle glaucoma.Investigative ophthalmology & visual science. 2011; 52(10):7122-7133. [PubMed]
- Braunger, B.M., R. Fuchshofer, and E.R. Tamm, The aqueous humor outflow pathways in glaucoma: A unifying concept of disease mechanisms and causative treatment.European Journal of Pharmaceutics and Biopharmaceutics. 2015; 95:173-181. [PubMed]
- Ryskamp, D.A., A.O. Jo, A.M. Frye, F. Vazquez-Chona, N. MacAulay, W.B. Thoreson, and D. Križaj, Swelling and eicosanoid metabolites differentially gate TRPV4 channels in retinal neurons and glia.Journal of Neuroscience. 2014; 34(47):15689-15700. [PubMed]
- Bennett, T.M., D.S. Mackay, C.J. Siegfried, and A. Shiels, Mutation of the melastatin-related cation channel, TRPM3, underlies inherited cataract and glaucoma.PloS one. 2014; 9(8):e104000. [PubMed]
- Liton, P.B., P. Gonzalez, and D.L. Epstein, The role of proteolytic cellular systems in trabecular meshwork homeostasis.Experimental eye research. 2009; 88(4):724-728. [PubMed]
- Rao, P.V., P.P. Pattabiraman, and C. Kopczynski, Role of the Rho GTPase/Rho kinase signaling pathway in pathogenesis and treatment of glaucoma: Bench to bedside research.Experimental eye research. 2017; 158:23-32. [PubMed]
- Leske, M.C., A. Heijl, M. Hussein, B. Bengtsson, L. Hyman, and E. Komaroff, Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial.Archives of ophthalmology. 2003; 121(1):48-56. [PubMed]
- Mozaffarieh, M. and J. Flammer, New insights in the pathogenesis and treatment of normal tension glaucoma.Current Opinion in Pharmacology. 2013; 13(1):43-49. [PubMed]
- Fingert, J.H., K. Miller, A. Hedberg-Buenz, B.R. Roos, C.J. Lewis, R.F. Mullins, and M.G. Anderson, Transgenic TBK1 mice have features of normal tension glaucoma.Human molecular genetics. 2016; 26(1):124-132. [PubMed]
- Kamal, D. and R. Hitchings, Normal tension glaucoma—a practical approach.British journal of ophthalmology. 1998; 82(7):835-840. [PubMed]
- Križaj, D., D.A. Ryskamp, N. Tian, G. Tezel, C.H. Mitchell, V.Z. Slepak, and V.I. Shestopalov, From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma.Current eye research. 2014; 39(2):105-119. [PubMed]
- Clark, A.F. and R.J. Wordinger, The role of steroids in outflow resistance.Experimental eye research. 2009; 88(4):752-759. [PubMed]
- Howell, G.R., R.T. Libby, T.C. Jakobs, R.S. Smith, F.C. Phalan, J.W. Barter, J.M. Barbay, J.K. Marchant, N. Mahesh, and V. Porciatti, Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma.J Cell Biol. 2007; 179(7):1523-1537. [PubMed]
- Whitmore, A.V., R.T. Libby, and S.W. John, Glaucoma: thinking in new ways—a role for autonomous axonal self-destruction and other compartmentalised processes?Progress in retinal and eye research. 2005; 24(6):639-662. [PubMed]
- Liu, B., S. McNally, J.I. Kilpatrick, S.P. Jarvis, and C.J. O’Brien, Aging and ocular tissue stiffness in glaucoma.Survey of ophthalmology. 2018; 63(1):56-74. [PubMed]
- Izzotti, A., M. Longobardi, C. Cartiglia, and S.C. Sacca, Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucoma.PLoS One. 2011; 6(1):e14567. [PubMed]
- Levkovitch-Verbin, H., S. Vander, D. Makarovsky, and F. Lavinsky, Increase in retinal ganglion cells’ susceptibility to elevated intraocular pressure and impairment of their endogenous neuroprotective mechanism by age.Molecular vision. 2013; 19:2011. [PubMed]
- Acott, T.S., M.J. Kelley, K.E. Keller, J.A. Vranka, D.W. Abu-Hassan, X. Li, M. Aga, and J.M. Bradley, Intraocular pressure homeostasis: maintaining balance in a high-pressure environment.Journal of Ocular Pharmacology and Therapeutics. 2014; 30(2-3):94-101. [PubMed]
- Liu, J.H., X. Zhang, D.F. Kripke, and R.N. Weinreb, Twenty-four-hour intraocular pressure pattern associated with early glaucomatous changes.Investigative ophthalmology & visual science. 2003; 44(4):1586-1590. [PubMed]
- Heijl, A., M.C. Leske, B. Bengtsson, L. Hyman, B. Bengtsson, and M. Hussein, Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial.Archives of ophthalmology. 2002; 120(10):1268-1279. [PubMed]
- Crooks, K.R., R.R. Allingham, X. Qin, Y. Liu, J.R. Gibson, C. Santiago-Turla, K.R. Larocque-Abramson, E. Del Bono, P. Challa, and L.W. Herndon, Genome-wide linkage scan for primary open angle glaucoma: influences of ancestry and age at diagnosis.PloS one. 2011; 6(7):e21967. [PubMed]
- Downs, J.C., IOP telemetry in the nonhuman primate.Exp Eye Res. 2015; 141:91-8. [PubMed]
- Agnifili, L., D. Pieragostino, A. Mastropasqua, V. Fasanella, L. Brescia, G.M. Tosi, P. Sacchetta, and L. Mastropasqua, Molecular biomarkers in primary open-angle glaucoma: from noninvasive to invasive, Volume 221, in Progress in brain research. 2015, Elsevier. p. 1-32. [PubMed]
- Cairns, E.A., W.H. Baldridge, and M.E. Kelly, The endocannabinoid system as a therapeutic target in glaucoma.Neural Plasticity. 2016; 2016. [PubMed]
- Stamer, W.D. and A.F. Clark, The many faces of the trabecular meshwork cell.Experimental eye research. 2017; 158:112-123. [PubMed]
- Ryskamp, D.A., A.M. Frye, T.T. Phuong, O. Yarishkin, A.O. Jo, Y. Xu, M. Lakk, A. Iuso, S.N. Redmon, and B. Ambati, TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye.Scientific reports. 2016; 6:30583. [PubMed]
- Grüsser, O.-J., U. Grüsser-Cornehls, R. Kusel, and A. Przybyszewski, Responses of retinal ganglion cells to eyeball deformation: a neurophysiological basis for “pressure phosphenes”.Vision research. 1989; 29(2):181-194. [PubMed]
- Kong, Y.X., J.G. Crowston, A.J. Vingrys, I.A. Trounce, and B.V. Bui, Functional changes in the retina during and after acute intraocular pressure elevation in mice.Investigative ophthalmology & visual science. 2009; 50(12):5732-5740. [PubMed]
- Ou, Y., R.E. Jo, E.M. Ullian, R.O. Wong, and L. Della Santina, Selective vulnerability of specific retinal ganglion cell types and synapses after transient ocular hypertension.Journal of Neuroscience. 2016; 36(35):9240-9252. [PubMed]
- Della Santina, L., D.M. Inman, C.B. Lupien, P.J. Horner, and R.O. Wong, Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma.Journal of Neuroscience. 2013; 33(44):17444-17457. [PubMed]
- Kong, L., M. Fry, M. Al-Samarraie, C. Gilbert, and P.G. Steinkuller, An update on progress and the changing epidemiology of causes of childhood blindness worldwide.Journal of American Association for Pediatric Ophthalmology and Strabismus. 2012; 16(6):501-507. [PubMed]
- Calkins, D.J., Critical pathogenic events underlying progression of neurodegeneration in glaucoma.Progress in retinal and eye research. 2012; 31(6):702-719. [PubMed]
- El-Danaf, R.N. and A.D. Huberman, Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types.Journal of Neuroscience. 2015; 35(6):2329-2343. [PubMed]
- Feng, L., H. Chen, J. Yi, J.B. Troy, H.F. Zhang, and X. Liu, Long-term protection of retinal ganglion cells and visual function by brain-derived neurotrophic factor in mice with ocular hypertension.Investigative ophthalmology & visual science. 2016; 57(8):3793-3802. [PubMed]
- Williams, P.A., J.R. Tribble, K.W. Pepper, S.D. Cross, B.P. Morgan, J.E. Morgan, S.W. John, and G.R. Howell, Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma.Molecular neurodegeneration. 2016; 11(1):26. [PubMed]
- Calkins, D.J., M. Pekny, M.L. Cooper, L. Benowitz, D. Calkins, M. Cooper, J. Crowston, A. Huberman, E. Johnson, and R. Lu, The challenge of regenerative therapies for the optic nerve in glaucoma.Experimental eye research. 2017; 157:28-33. [PubMed]
- Beirowski, B., E. Babetto, M.P. Coleman, and K.R. Martin, The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model.European Journal of Neuroscience. 2008; 28(6):1166-1179. [PubMed]
- Quigley, H.A., R.M. Sanchez, G.R. Dunkelberger, N.L. L’Hernault, and T.A. Baginski, Chronic glaucoma selectively damages large optic nerve fibers.Investigative Ophthalmology & Visual Science. 1987; 28(6):913-920. [PubMed]
- Morgan, J.E., Selective cell death in glaucoma: does it really occur? The British journal of ophthalmology. 1994; 78(11):875. [PubMed]
- Jakobs, T.C., R.T. Libby, Y. Ben, S.W. John, and R.H. Masland, Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005; 171(2):313-325. [PubMed]
- Feng, L., Y. Zhao, M. Yoshida, H. Chen, J.F. Yang, T.S. Kim, J. Cang, J.B. Troy, and X. Liu, Sustained ocular hypertension induces dendritic degeneration of mouse retinal ganglion cells that depends on cell type and location.Investigative ophthalmology & visual science. 2013; 54(2):1106-1117. [PubMed]
- Risner, M.L., S. Pasini, M.L. Cooper, W.S. Lambert, and D.J. Calkins, Axogenic mechanism enhances retinal ganglion cell excitability during early progression in glaucoma.Proceedings of the National Academy of Sciences. 2018; 115(10):E2393-E2402. [PubMed]
- Križaj, D., Polymodal sensory integration in retinal ganglion cells, in Retinal Degenerative Diseases. 2016, Springer. p. 693-698. [PubMed]
- de Sevilla Müller, L.P., A. Sargoy, A.R. Rodriguez, and N.C. Brecha, Melanopsin ganglion cells are the most resistant retinal ganglion cell type to axonal injury in the rat retina.PLoS One. 2014; 9(3):e93274. [PubMed]
- Lakk, M., D. Young, J.M. Baumann, A.O. Jo, H. Hu, and D. Križaj, Polymodal TRPV1 and TRPV4 sensors colocalize but do not functionally interact in a subpopulation of mouse retinal ganglion cells.Frontiers in cellular neuroscience. 2018; 12. [PubMed]
- Huang, W., J.B. Fileta, A. Dobberfuhl, T. Filippopolous, Y. Guo, G. Kwon, and C.L. Grosskreutz, Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma.Proceedings of the National Academy of Sciences. 2005; 102(34):12242-12247. [PubMed]
- Huang, W., W. Xing, D.A. Ryskamp, C. Punzo, and D. Križaj, Localization and phenotype-specific expression of ryanodine calcium release channels in C57BL6 and DBA/2J mouse strains.Experimental eye research. 2011; 93(5):700-709. [PubMed]
- Kroeger, H., W.C. Chiang, J. Felden, A. Nguyen, and J.H. Lin, ER stress and unfolded protein response in ocular health and disease.The FEBS journal. 2019; 286(2):399-412. [PubMed]
- Guymer, C., J.P. Wood, G. Chidlow, and R.J. Casson, Neuroprotection in glaucoma: recent advances and clinical translation.Clinical & experimental ophthalmology. 2019; 47(1):88-105. [PubMed]
- Hare, W.A., E. WoldeMussie, R.N. Weinreb, H. Ton, G. Ruiz, M. Wijono, B. Feldmann, L. Zangwill, and L. Wheeler, Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey, II: Structural measures.Investigative ophthalmology & visual science. 2004; 45(8):2640-2651. [PubMed]
- Rutar, M., R. Natoli, R. Albarracin, K. Valter, and J. Provis, 670-nm light treatment reduces complement propagation following retinal degeneration.J Neuroinflammation. 2012; 9:257. [PubMed]
- Lim, J.-H.A., B.K. Stafford, P.L. Nguyen, B.V. Lien, C. Wang, K. Zukor, Z. He, and A.D. Huberman, Neural activity promotes long-distance, target-specific regeneration of adult retinal axons.Nature neuroscience. 2016; 19(8):1073. [PubMed]
- Yang, H., J. Reynaud, H. Lockwood, G. Williams, C. Hardin, L. Reyes, C. Stowell, S.K. Gardiner, and C.F. Burgoyne, The connective tissue phenotype of glaucomatous cupping in the monkey eye-clinical and research implications.Progress in retinal and eye research. 2017; 59:1-52. [PubMed]
- Eilaghi, A., J.G. Flanagan, C.A. Simmons, and C.R. Ethier, Effects of scleral stiffness properties on optic nerve head biomechanics.Annals of biomedical engineering. 2010; 38(4):1586-1592. [PubMed]
- Burgoyne, C.F. and J.C. Downs, Premise and prediction–how optic nerve head biomechanics underlies the susceptibility and clinical behavior of the aged optic nerve head.Journal of glaucoma. 2008; 17(4):318. [PubMed]
- Howell, G.R., I. Soto, X. Zhu, M. Ryan, D.G. Macalinao, G.L. Sousa, L.B. Caddle, K.H. MacNicoll, J.M. Barbay, and V. Porciatti, Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma.The Journal of clinical investigation. 2012; 122(4):1246-1261. [PubMed]
- Osborne, N.N., C. Núñez-Álvarez, B. Joglar, and S. del Olmo-Aguado, Glaucoma: focus on mitochondria in relation to pathogenesis and neuroprotection.European journal of pharmacology. 2016; 787:127-133. [PubMed]
- Dengler-Crish, C.M., M.A. Smith, D.M. Inman, G.N. Wilson, J.W. Young, and S.D. Crish, Anterograde transport blockade precedes deficits in retrograde transport in the visual projection of the DBA/2J mouse model of glaucoma.Frontiers in neuroscience. 2014; 8:290. [PubMed]
- Bosco, A., M.R. Steele, and M.L. Vetter, Early microglia activation in a mouse model of chronic glaucoma.Journal of Comparative Neurology. 2011; 519(4):599-620. [PubMed]
- Inman, D.M. and P.J. Horner, Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma.Glia. 2007; 55(9):942-953. [PubMed]
- Sun, D., M. Lye‐Barthel, R.H. Masland, and T.C. Jakobs, The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse.Journal of Comparative Neurology. 2009; 516(1):1-19. [PubMed]
- Ly, T., N. Gupta, R.N. Weinreb, P.L. Kaufman, and Y.H. Yücel, Dendrite plasticity in the lateral geniculate nucleus in primate glaucoma.Vision research. 2011; 51(2):243-250. [PubMed]
- Gupta, N., G. Greenberg, L.N. De Tilly, B. Gray, M. Polemidiotis, and Y.H. Yücel, Atrophy of the lateral geniculate nucleus in human glaucoma detected by magnetic resonance imaging.British Journal of Ophthalmology. 2009; 93(1):56-60. [PubMed]
- Geeraerts, E., M. Claes, E. Dekeyster, M. Salinas-Navarro, L. De Groef, C. Van den Haute, I. Scheyltjens, V. Baekelandt, L. Arckens, and L. Moons, Optogenetic stimulation of the superior colliculus confers retinal neuroprotection in a mouse glaucoma model.Journal of Neuroscience. 2019; 39(12):2313-2325. [PubMed]
- Coulombre, A.J., Cytology of the developing eye, Volume 11, in International review of cytology. 1961, Elsevier. p. 161-194. [PubMed]
- Shelton, L., D. Troilo, M.R. Lerner, Y. Gusev, D.J. Brackett, and J.S. Rada, Microarray analysis of choroid/RPE gene expression in marmoset eyes undergoing changes in ocular growth and refraction.Molecular vision. 2008; 14:1465. [PubMed]
- Schneider, M. and R. Fuchshofer, The role of astrocytes in optic nerve head fibrosis in glaucoma.Experimental eye research. 2016; 142:49-55. [PubMed]
- Tamm, E. and C. Ethier, Lasker/IRRF Initiative on Astrocytes and Glaucomatous Neurodegeneration Participants. Biological aspects of axonal damage in glaucoma: a brief review.Exp Eye Res. 2017; 157:5-12. [PubMed]
- Jakobs, T.C., Differential gene expression in glaucoma.Cold Spring Harbor perspectives in medicine. 2014; 4(7):a020636. [PubMed]
- Tovar-Vidales, T., R.J. Wordinger, and A.F. Clark, Identification and localization of lamina cribrosa cells in the human optic nerve head.Experimental eye research. 2016; 147:94-97. [PubMed]
- Mansour, H., J.R. McColm, L. Cole, M. Weible II, A. Korlimbinis, and T. Chan-Ling, Connexin 30 expression and frequency of connexin heterogeneity in astrocyte gap junction plaques increase with age in the rat retina.PloS one. 2013; 8(3):e57038. [PubMed]
- Newman, E.A., Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters.Philosophical Transactions of the Royal Society B: Biological Sciences. 2015; 370(1672):20140195. [PubMed]
- Hernandez, M.R., The optic nerve head in glaucoma: role of astrocytes in tissue remodeling.Progress in retinal and eye research. 2000; 19(3):297-321. [PubMed]
- Lye-Barthel, M., D. Sun, and T.C. Jakobs, Morphology of astrocytes in a glaucomatous optic nerve.Investigative ophthalmology & visual science. 2013; 54(2):909-917. [PubMed]
- Dvoriantchikova, G. and D. Ivanov, Tumor necrosis factor‐alpha mediates activation of NF‐κB and JNK signaling cascades in retinal ganglion cells and astrocytes in opposite ways.European Journal of Neuroscience. 2014; 40(8):3171-3178. [PubMed]
- Johnson, E.C., L. Jia, W.O. Cepurna, T.A. Doser, and J.C. Morrison, Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model.Investigative ophthalmology & visual science. 2007; 48(7):3161-3177. [PubMed]
- Tezel, G., M.R. Hernandez, and M.B. Wax, In vitro evaluation of reactive astrocyte migration, a component of tissue remodeling in glaucomatous optic nerve head.Glia. 2001; 34(3):178-189. [PubMed]
- Redmon, S., O. Yarishkin, M. Lakk, and D. Krizaj, Retinal glial cells respond differently to the activation of the mechanosensitive channel TRPV4.Investigative Ophthalmology & Visual Science. 2018; 59(9):3939-3939.
- Chong, R.S. and K.R. Martin, Glial cell interactions and glaucoma.Current opinion in ophthalmology. 2015; 26(2):73. [PubMed]
- Wax, M.B. and G. Tezel, Immunoregulation of retinal ganglion cell fate in glaucoma.Experimental Eye Research. 2009; 88(4):825-830. [PubMed]
- Anderson, S.R., J. Zhang, M.R. Steele, C.O. Romero, A.G. Kautzman, D.P. Schafer, and M.L. Vetter, Complement targets newborn retinal ganglion cells for phagocytic elimination by microglia.Journal of Neuroscience. 2019; 39(11):2025-2040. [PubMed]
- Breen, K.T., S.R. Anderson, M.R. Steele, D.J. Calkins, A. Bosco, and M.L. Vetter, Loss of fractalkine signaling exacerbates axon transport dysfunction in a chronic model of glaucoma.Frontiers in neuroscience. 2016; 10:526. [PubMed]
- Wang, M., X. Wang, L. Zhao, W. Ma, I.R. Rodriguez, R.N. Fariss, and W.T. Wong, Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina.Journal of Neuroscience. 2014; 34(10):3793-3806. [PubMed]
- Bosco, A., C.O. Romero, K.T. Breen, A.A. Chagovetz, M.R. Steele, B.K. Ambati, and M.L. Vetter, Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma.Disease models & mechanisms. 2015; 8(5):443-455. [PubMed]
- Karlstetter, M., R. Scholz, M. Rutar, W.T. Wong, J.M. Provis, and T. Langmann, Retinal microglia: just bystander or target for therapy?Progress in retinal and eye research. 2015; 45:30-57. [PubMed]
- Matsumoto, H., S. Sugio, F. Seghers, D. Krizaj, H. Akiyama, Y. Ishizaki, P. Gailly, and K. Shibasaki, Retinal detachment-induced Müller glial cell swelling activates TRPV4 ion channels and triggers photoreceptor death at body temperature.Journal of Neuroscience. 2018; 38(41):8745-8758. [PubMed]
- Reichenbach, A. and A. Bringmann, Müller cells in the healthy and diseased retina. Springer Science & Business Media. 2010, New York: Springer Verlag. [PubMed]
- Jo, A.O., D.A. Ryskamp, T.T. Phuong, A.S. Verkman, O. Yarishkin, N. MacAulay, and D. Križaj, TRPV4 and AQP4 channels synergistically regulate cell volume and calcium homeostasis in retinal Müller glia.Journal of Neuroscience. 2015; 35(39):13525-13537. [PubMed]
- Nawashiro, H., A. Messing, N. Azzam, and M. Brenner, Mice lacking GFAP are hypersensitive to traumatic cerebrospinal injury.Neuroreport. 1998; 9(8):1691-1696. [PubMed]
- Morrison, J.C., W.O.C.Y. Guo, and E.C. Johnson, Pathophysiology of human glaucomatous optic nerve damage: insights from rodent models of glaucoma.Experimental eye research. 2011; 93(2):156-164. [PubMed]
- Vargas, J.L.C., I.K. Osswald, N. Unsain, M.R. Aurousseau, P.A. Barker, D. Bowie, and A. Di Polo, Soluble tumor necrosis factor alpha promotes retinal ganglion cell death in glaucoma via calcium-permeable AMPA receptor activation.Journal of Neuroscience. 2015; 35(35):12088-12102. [PubMed]
- Jassim, A.H. and D.M. Inman, Evidence of Hypoxic Glial Cells in a Model of Ocular Hypertension.Investigative Ophthalmology & Visual Science. 2019; 60(1):1-15. [PubMed]
- Jo, A.O., M. Lakk, A.M. Frye, T.T. Phuong, S.N. Redmon, R. Roberts, B.A. Berkowitz, O. Yarishkin, and D. Križaj, Differential volume regulation and calcium signaling in two ciliary body cell types is subserved by TRPV4 channels.Proceedings of the National Academy of Sciences. 2016; 113(14):3885-3890. [PubMed]
- Križaj, D., No cell is an island: trabecular meshwork ion channels as sensors of the ambient milieu, Volume 3, in Glaucoma Research and Clinical Advances J.R. Samples and P.A. Knepper, Editors. 2019, Kugler Publications Amsterdam.
- Lindqvist, N., Q. Liu, J. Zajadacz, K. Franze, and A. Reichenbach, Retinal glial (Müller) cells: sensing and responding to tissue stretch.Investigative ophthalmology & visual science. 2010; 51(3):1683-1690. [PubMed]
- Niittykoski, M., G. Kalesnykas, K.P. Larsson, K. Kaarniranta, K.E. Åkerman, and H. Uusitalo, Altered calcium signaling in an experimental model of glaucoma.Investigative ophthalmology & visual science. 2010; 51(12):6387-6393. [PubMed]
- Ribas, V.T., J.C. Koch, U. Michel, M. Bähr, and P. Lingor, Attenuation of axonal degeneration by calcium channel inhibitors improves retinal ganglion cell survival and regeneration after optic nerve crush.Molecular neurobiology. 2017; 54(1):72-86. [PubMed]
- Ingber, D.E., Tensegrity: the architectural basis of cellular mechanotransduction.Annual review of physiology. 1997; 59(1):575-599. [PubMed]
- Ranade, S.S., R. Syeda, and A. Patapoutian, Mechanically activated ion channels.Neuron. 2015; 87(6):1162-1179. [PubMed]
- Jaalouk, D.E. and J. Lammerding, Mechanotransduction gone awry.Nature reviews Molecular cell biology. 2009; 10(1):63. [PubMed]
- Cox, C.D., N. Bavi, and B. Martinac, Origin of the force: the force-from-lipids principle applied to piezo channels, Volume79, in Current topics in membranes. 2017, Elsevier. p. 59-96. [PubMed]
- Phuong, T.T., S.N. Redmon, O. Yarishkin, J.M. Winter, D.Y. Li, and D. Križaj, Calcium influx through TRPV4 channels modulates the adherens contacts between retinal microvascular endothelial cells.The Journal of physiology. 2017; 595(22):6869-6885. [PubMed]
- Sappington, R.M., T. Sidorova, N.J. Ward, R. Chakravarthy, K.W. Ho, and D.J. Calkins, Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress.Channels. 2015; 9(2):102-113. [PubMed]
- Yarishkin, O., T.T. Phuong, C.A. Bretz, K.W. Olsen, J.M. Baumann, M. Lakk, A. Crandall, C. Heurteaux, M.E. Hartnett, and D. Križaj, TREK-1 channels regulate pressure sensitivity and calcium signaling in trabecular meshwork cells.The Journal of general physiology. 2018; 150(12):1660-1675. [PubMed]
- Redmon, S., N., K. Shibasaki, and D. Križaj,Transient receptor potential cation channel subfamily V member 4, in Encyclopedia of Signaling Molecules, 2nd Edition, S. Choi, Editor. 2017, Springer Press.
- White, J.P., M. Cibelli, L. Urban, B. Nilius, J.G. McGeown, and I. Nagy, TRPV4: molecular conductor of a diverse orchestra.Physiological reviews. 2016; 96(3):911-973. [PubMed]
- Lapajne, L., M. Lakk, L. Gubeljak, O. Yarishkin, M. Hawlina, and D. Križaj, Corneal mechanotransduction drives TRPV4-dependent release of the transmitter ATP.Investigative ophthalmology & visual science. 2019; 60, Suppl..
- Ryskamp, D.A., P. Witkovsky, P. Barabas, W. Huang, C. Koehler, N.P. Akimov, S.H. Lee, S. Chauhan, W. Xing, and R.C. Rentería, The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells.Journal of Neuroscience. 2011; 31(19):7089-7101. [PubMed]
- Arredondo, D.Z., R.I. Noguez, A.C. Bautista, O.R. Vázquez, M. Bernardini, A.P. Fiorio, D. Gkika, N. Prevarskaya, F. Lopez-Casillas, and W. Liedtke, Dual contribution of TRPV4 antagonism in the regulatory effect of vasoinhibins on blood-retinal barrier permeability: diabetic milieu makes a difference.Scientific reports. 2017; 7(1):13094-13094. [PubMed]
- Lakk, M., O. Yarishkin, J.M. Baumann, A. Iuso, and D. Križaj, Cholesterol regulates polymodal sensory transduction in Müller glia.Glia. 2017; 65(12):2038-2050. [PubMed]
- Alloui, A., K. Zimmermann, J. Mamet, F. Duprat, J. Noel, J. Chemin, N. Guy, N. Blondeau, N. Voilley, and C. Rubat‐Coudert,TREK‐1, a K+ channel involved in polymodal pain perception.The EMBO journal. 2006; 25(11):2368-2376. [PubMed]
- Harrington, D. and M. Drake, The visual fields: text and atlas of clinical perimetry. 6th. 1990, Mosby: Philadelphia. p. 52-60.
- Bonomi, L., G. Marchini, M. Marraffa, and R. Morbio, The relationship between intraocular pressure and glaucoma in a defined population.Ophthalmologica. 2001; 215(1):34-38. [PubMed]
- Vecino, E., F.D. Rodriguez, N. Ruzafa, X. Pereiro, and S.C. Sharma, Glia–neuron interactions in the mammalian retina.Progress in retinal and eye research. 2016; 51:1-40. [PubMed]
- Samuels, B.C., J.T. Siegwart, W. Zhan, L. Hethcox, M. Chimento, R. Whitley, J.C. Downs, and C.A. Girkin, A Novel Tree Shrew (Tupaia belangeri) Model of Glaucoma.Investigative ophthalmology & visual science. 2018; 59(7):3136-3143. [PubMed]
- Tamm, E.R., C.R. Ethier, J.E. Dowling, C. Downs, M.H. Ellisman, S. Fisher, B. Fortune, M. Fruttiger, T. Jakobs, and G. Lewis, Biological aspects of axonal damage in glaucoma: a brief review.Experimental eye research. 2017; 157:5-12. [PubMed]
- Lieff, J. Searching for the Mind.2013; Available from: http://jonlieffmd.com/blog/are-microglia-the-most-intelligent-brain-cells. [PubMed]
- Moran, M.M., M.A. McAlexander, T. Bíró, and A. Szallasi, Transient receptor potential channels as therapeutic targets.Nature reviews Drug discovery. 2011; 10(8):601. [PubMed]
- Patel, A.J. and E. Honoré, Properties and modulation of mammalian 2P domain K+ channels.Trends in neurosciences. 2001; 24(6):339-346. [PubMed]
- Yarishkin, O., T.T. Phuong, M. Lakk, and D. Križaj, TRPV4 does not regulate the distal retinal light response, in Retinal Degenerative Diseases. 2018, Springer. p. 553-560. [PubMed]